WO1999064002A1 - Spiropiperidine derivatives as melanocortin receptor agonists - Google Patents
Spiropiperidine derivatives as melanocortin receptor agonists Download PDFInfo
- Publication number
- WO1999064002A1 WO1999064002A1 PCT/US1999/013252 US9913252W WO9964002A1 WO 1999064002 A1 WO1999064002 A1 WO 1999064002A1 US 9913252 W US9913252 W US 9913252W WO 9964002 A1 WO9964002 A1 WO 9964002A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- treatment
- prevention
- formula
- heteroaryl
- Prior art date
Links
- 0 C(*1)C2(CCNCC2)c2c1cccc2 Chemical compound C(*1)C2(CCNCC2)c2c1cccc2 0.000 description 5
- OTLZOGPZTLEBPO-HNNXBMFYSA-N CC(C)(C)OC(N(CC1)CCC1(C1)c2ccccc2[C@H]1C(CO)=O)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1(C1)c2ccccc2[C@H]1C(CO)=O)=O OTLZOGPZTLEBPO-HNNXBMFYSA-N 0.000 description 1
- PAYURZPMFLHXGU-UHFFFAOYSA-N CC(C)(C)OC(N(CCCC1)CCCc2ccccc2C1=C(O)O)=O Chemical compound CC(C)(C)OC(N(CCCC1)CCCc2ccccc2C1=C(O)O)=O PAYURZPMFLHXGU-UHFFFAOYSA-N 0.000 description 1
- LBEXLSMEKCNRAB-UHFFFAOYSA-N OC(C(C1)C(CCCC2)C2C1(CC1)CCN1I)=O Chemical compound OC(C(C1)C(CCCC2)C2C1(CC1)CCN1I)=O LBEXLSMEKCNRAB-UHFFFAOYSA-N 0.000 description 1
- RLDFRVFBWQJHTC-UHFFFAOYSA-N OC(C(C1)c2ccccc2C1(CC1)CCN1I)=O Chemical compound OC(C(C1)c2ccccc2C1(CC1)CCN1I)=O RLDFRVFBWQJHTC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06191—Dipeptides containing heteroatoms different from O, S, or N
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Definitions
- Spiropiperidine derivatives are melanocortin receptor agonists, and as such are useful in the treatment of disorders responsive to the activation of melanocortin receptors, such as obesity, diabetes as well as male and/or female sexual dysfunction.
- Pro-opiomelanocortin (POMC) derived peptides are known to affect food intake.
- GPCRs G-protein coupled receptors
- M-R melanocortin receptor
- MC-Rs evidence for the involvement of MC-Rs in obesity includes: i) the agouti (A ) mouse which ectopically expresses an antagonist of the MC-1R, MC-3R and -4R is obese, indicating that blocking the action of these three MC-Rs can lead to hyperphagia and metabolic disorders; ii) MC-4R knockout mice (Huszar et al., Cell, 88, 131-141, 1997) recapitulate the phenotype of the agouti mouse and these mice are obese; iii) the cyclic heptapeptide MT-II (MC-1R, -3R, -4R, -5R, agonist) injected intracerebroventricularly (ICV) in rodents, reduces food intake in several animal feeding models (NPY, oh/oh, agouti, fasted) while ICV injected SHU-9119 (MC-3R, -4R antagonist; MC-1R and -5R
- MC-1R Five MC-Rs have thus far been identified, and these are expressed in different tissues.
- MC-1R was initially characterized by dominant gain of function mutations at the Extension locus, affecting coat color by controlling phaeomelanin to eumelanin conversion through control of tyrosinase.
- MC-1R is mainly expressed in melanocytes.
- MC-2R is expressed in the adrenal gland and represents the ACTH receptor.
- MC-3R is expressed in the brain, gut and placenta and may be involved in the control of food intake and thermogenesis.
- MC-4R is uniquely expressed in the brain and its inactivation was shown to cause obesity.
- MC-5R is expressed in many tissues including white fat, placenta and exocrine glands. A low level of expression is also observed in the brain.
- MC-5R knock out mice reveal reduced sebaceous gland lipid production (Chen et al., Cell, 1997, 91, 789-798).
- MT-II melanotan -II
- Intramuscular administration of MT-II (0.025 mg/kg and 0.1 mg/kg) to 10 non-organic impotent patients caused transient erections (8 responders) with onset from 50-180 minutes; penile erections subsided after ejaculation (15th American Peptide Symposium 6/14-19, 1997, Nashville, TN, study now published in J. Urology, 160, 389-393, 1998).
- the present invention provides compounds having the formula I:
- Cy2 is . a six-membered aromatic ring containing 0 or 1 N atom or cyclohexane
- X is O, CH2, SO2, CHCO2RD, CHSO2R , CHC(O)N(Rb) , NRb,
- NSO2R a NSO2N(Rb)2, NCORa, NCON(Rb) 2 , CHN(Rb)COR a , CHN(Rb)SO2R a , CHCH2ORb, or CH(CH2)-heteroaryl
- Rl is H, Ci- ⁇ alkyl, CH(Rb)-aryl, CH(Rb)-heteroaryl, (CH2) n -
- R2 is H or halo
- Ra is Rb, (CH2) n N(Rb)2, (CH2) n NH-2- pyridyl, (CH2) n NH-2-imidazolyl, (CH2) n NH-2-thiazolyl, (CH2) n NH-2-pyrimidinyl,
- Rb is H, Ci-8alkyl, (CH2) n aryl, (CH2) n heteroaryl, C3-6cycloalkyl; or 2 Rb together with the nitrogen atom to which they are attached form a 5- or 6-membered ring optionally containing an additional heteroatom selected from 0, S, and NRl ;
- Rc is Rb, halo, ORb, NHSO2R , N(Rb) 2 , CN, NO2, SO2N(Rb)2, SO2R ,
- Rd is H, NO2, or CN
- Cy is aryl, 5- or 6-membered heteroaryl, 5- or 6-membered heterocyclyl, or
- n is 0 to 3 ; m, p and q are independently 0, 1 or 2; r is 1 , 2 or 3 ; or a pharmaceutically acceptable salt thereof.
- compounds of formula I are compounds wherein Cy2 is benzene or cyclohexane.
- compounds of formula I are compounds wherein X is CHCO2R b , CHC(O)N(Rb)2, NSO2R a CHN(Rb)COR a , CHN(Rb)SO2R a CHCH2ORb or CHCH2-heteroaryl.
- Rb and R c are as defined under formula I, and Cy is aryl, 5- or 6-membered heteroaryl, or 5-or 6-membered carbocyclyl.
- Cy is benzene or cyclohexane.
- X is CHCO2Rb, CHC(O)N(Rb)2, NSO2R a CHN(Rb)CORa, or
- R2 is H or halo
- Ra is Rb, (CH2) n N(R )2, (CH2) n NH-2-pyridyl, (CH2) n H-2-imidazolyl,
- Rb is H, C i - ⁇ alkyl, (CH2)naryl, (CH2)nheteroaryl, or C3-6cycloalkyl;
- Cy is benzene, pyridine, imidazole or cyclohexane; nis 0to3; or a pharmaceutically acceptable salt thereof.
- R is H, Ci-8alkyl, (CH2)naryl, (CH2)nheteroaryl, or C3-6cycloalkyl;
- Rc is H, halo, Rb, ORb, CF3, OCF3;
- Cyis benzene pyridine, imidazole or cyclohexane; nis 0to3; or a pharmaceutically acceptable salt thereof.
- the carbon atom marked with * has the R configuration.
- Cy is benzene or cyclohexane.
- Another aspect of the present invention provides a method for the treatment or prevention of obesity or diabetes in a mammal which comprises administering to said mammal an effective amount of a compound of formula I.
- Another aspect of the present invention provides a method for the treatment or prevention of male or female sexual dysfunction including erectile dysfunction which comprises administering to a patient in need of such treatment or prevention an effective amount of a compound of formula I.
- Another aspect of the present invention provides a method for the treatment or prevention of male or female sexual dysfunction including erectile dysfunction which comprises administering to a patient in need of such treatment or prevention an effective amount of an agonist of melanocortin-4 receptor.
- Yet another aspect of the present invention provides a pharmaceutical composition comprising a compound of formula I and a pharmaceutically acceptable carrier.
- alkyl groups specified above are intended to include those alkyl groups of the designated length in either a straight or branched configuration. Exemplary of such alkyl groups are methyl, ethyl, propyl, isopropyl. butyl, sec-butyl, tertiary butyl, pentyl, isopentyl, hexyl, isohexyl, and the like.
- halogen is intended to include the halogen atoms fluorine, chlorine, bromine and iodine.
- aryl includes phenyl and naphthyl.
- heteroaryl includes mono- and bicyclic aromatic rings containing from 1 to 4 heteroatoms selected from nitrogen, oxygen and sulfur.
- "5- or 6-membered heteroaryl” are monocyclic heteroaromatic rings, examples thereof include thiazole. oxazole, thiophene, furan, pyrrole, imidazole, isoxazole, pyrazole, triazole, thiadiazole, tetrazole, oxadiazole, pyridine, pyridazine, pyrazine, and the like.
- Bicyclic heteroaromatic rings include, but are not limited to, benzothiadiazole.
- indole benzothiophene, benzofuran, benzimidazole, benzisoxazole, benzothiazole, quinoline, benzotriazole, benzoxazole, isoquinoline.
- purine furopyridine and thienopyridine.
- 5- or 6-membered carbocyclyl is intended to include non- aromatic rings containing only carbon atoms such as cyclopentyl and cyclohexyl.
- 5 and 6-membered heterocyclyl is intended to include non- aromatic heterocvcles containing one to four heteroatoms selected from nitrogen, oxygen and sulfur.
- Examples of a 5 or 6-membered heterocyclyl include piperidine, morpholine, thiamorpholine, pyrrolidine, imidazolidine, tetrahydrofuran, piperazine, and the like.
- NR b Rb may represent NH2, NHCH3, N(CH3)CH2CH3, and the like.
- composition as in pharmaceutical composition, is intended to encompass a product comprising the active ingredient(s), and the inert ingredient(s) that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the present invention and a pharmaceutically acceptable carrier.
- Erectile dysfunction is a disorder involving the failure of a male mammal to achieve erection, ejaculation, or both. Symptoms of erectile dysfunction include an inability to achieve or maintain an erection, ejaculatory failure, premature ejaculation, inability to achieve an orgasm. An increase in erectile dysfunction is often associated with age and is generally caused by a physical disease or as a side-effect of drug treatment.
- Female sexual dysfunction encompasses, without limitatin. conditions usch as a lack of sexual desire and related arousal disorders, inhibited orgasm, lubrication difficulties, and vaginismus.
- Compounds of Formula I contain one or more asymmetric centers and can thus occur as racemates and racemic mixtures, single enantiomers, diastereomeric mixtures and individual diastereomers.
- the present invention is meant to comprehend all such isomeric forms of the compounds of Formula I.
- Some of the compounds described herein contain olefmic double bonds, and unless specified otherwise, are meant to include both E and Z geometric isomers.
- Some of the compounds described herein may exist as tautomers such as keto-enol tautomers.
- the individual tautomers as well as mixture thereof are encompassed with compounds of Formula I.
- Compounds of the Formula I may be separated into diastereoisomeric pairs of enantiomers by, for example, fractional crystallization from a suitable solvent, for example methanol or ethyl acetate or a mixture thereof.
- a suitable solvent for example methanol or ethyl acetate or a mixture thereof.
- the pair of enantiomers thus obtained may be separated into individual stereoisomers by conventional means, for example by the use of an optically active acid as a resolving agent.
- any enantiomer of a compound of the general Formula I or la may be obtained by stereospecific synthesis using optically pure starting materials or reagents of known configuration.
- salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids.
- Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, lithium, magnesium, potassium, and sodium salts.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl-morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- basic ion exchange resins such as
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, formic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, malonic, mucic, nitric, pamoic, pantothenic, phosphoric, propionic, succinic, sulfuric, tartaric, p-toluenesulfonic acid, trifluoroacetic acid, and the like.
- Particularly preferred are citric, fumaric, hydrobromic, hydrochloric, maleic, phosphoric, sulfuric, and tartaric acids.
- Compounds of formula I are melanocortin receptor agonists and as such are useful in the treatment, control or prevention of diseases, disorders or conditions responsive to the activation of one or more of the melanocortin receptors including, but are not limited to, MC-1, MC-2, MC-3, MC-4, or MC-5.
- Such diseases, disorders or conditions include, but are not limited to, obesity (by reducing appetite, increasing metabolic rate, reducing fat intake or reducing carbohydrate craving), diabetes mellitus (by enhancing glucose tolerance, decreasing insulin resistance), hypertension, hyperlipidemia, osteoarthritis, cancer, gall bladder disease, sleep apnea, depression, anxiety, compulsion, neuroses, insomnia/sleep disorder, substance abuse, pain, male and female sexual dysfunction (including impotence, loss of libido and erectile dysfunction), fever, inflammation, immune modulation, rheumatoid arthritis, skin tanning, acne and other skin disorders, neuroprotective and cognitive and memory enhancement including the treatment of Alzheimer's disease.
- Some compounds of formula I show highly specific activity toward the melanocortin- 4 receptor which makes them especially useful in the prevention and treatment of obesity, as well as male and female sexual dysfunction.
- Any suitable route of administration may be employed for providing a mammal, especially a human with an effective dosage of a compound of the present invention.
- oral, rectal, topical, parenteral, ocular, pulmonary, nasal, and the like may be employed.
- Dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols, and the like.
- compounds of Formula I are administered orally.
- the effective dosage of active ingredient employed may vary depending on the particular compound employed, the mode of administration, the condition being treated and the severity of the condition being treated. Such dosage may be ascertained readily by a person skilled in the art.
- the compounds of the present invention are administered at a daily dosage of from 0.01 milligram to about 100 milligrams per kilogram of animal body weight, preferably given in a single dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dose will generally be from about 0.7 milligrams to about 3500 milligrams. This dosage regimen may be adjusted to provide the optimal therapeutic response.
- the compounds of the present invention are administered at a daily dosage of from about 0.001 milligram to about 100 milligram per kilogram of animal body weight, preferably given in a single dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dose will generally be from about 0.07 milligrams to about 350 milligrams. This dosage regimen may be adjusted to provide the optimal therapeutic response.
- sexual dysfunction compounds of the present invention are given in a dose range of 0.001 milligram to about 100 milligram per kilogram of body weight, preferably as a single dose orally or as a nasal spray.
- compositions which comprises a compound of Formula I and a pharmaceutically acceptable carrier.
- the pharmaceutical compositions of the present invention comprise a compound of Formula I as an active ingredient or a pharmaceutically acceptable salt thereof, and may also contain a pharmaceutically acceptable carrier and optionally other therapeutic ingredients.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic bases or acids and organic bases or acids.
- the compounds of Formula I can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques.
- the carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g., oral or parenteral (including intravenous).
- any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents and the like in the case of oral liquid preparations, such as.
- oral solid preparations such as, for example, powders, hard and soft capsules and tablets, with the solid oral preparations being preferred over the liquid preparations.
- tablets and capsules represent the most advantageous oral dosage unit form in which case solid pharmaceutical carriers are obviously employed. If desired, tablets may be coated by standard aqueous or nonaqueous techniques. Such compositions and preparations should contain at least 0.1 percent of active compound. The percentage of active compound in these compositions may, of course, be varied and may conveniently be between about 2 percent to about 60 percent of the weight of the unit. The amount of active compound in such therapeutically useful compositions is such that an effective dosage will be obtained.
- the active compounds can also be administered intranasally as, for example, liquid drops or spray.
- the tablets, pills, capsules, and the like may also contain a binder such as gum tragacanth, acacia, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid; a lubricant such as magnesium stearate; and a sweetening agent such as sucrose, lactose or saccharin.
- a dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier such as a fatty oil.
- tablets may be coated with shellac, sugar or both.
- a syrup or elixir may contain, in addition to the active ingredient, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and a flavoring such as cherry or orange flavor.
- Compounds of formula I may also be administered parenterally.
- Solutions or suspensions of these active compounds can be prepared in water suitably mixed with a surfactant such as hydroxy-propylcellulose.
- Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
- the pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions.
- the form must be sterile and must be fluid to the extent that easy syringability exists. It must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol
- Combination Therapy Compounds of Formula I may be used in combination with other drugs that are used in the treatment/prevention/suppression or amelioration of the diseases or conditions for which compounds of Formula I are useful. Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of Formula I.
- a pharmaceutical composition containing such other drugs in addition to the compound of Formula I is preferred.
- the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of Formula I. Examples of other active ingredients that may be combined with a compound of Formula I, either administered separately or in the same pharmaceutical compositions, include, but are not limited to:
- insulin sensitizers including (i) PPAR ⁇ agonists such as the glitazones (e.g. troglitazone, pioglitazone, englitazone, MCC-555. BRL49653 and the like), and compounds disclosed in WO97/27857, 97/28115, 97/28137 and 97/27847; (ii) biguanides such as metformin and phenformin;
- ⁇ -glucosidase inhibitors such as acarbose
- e cholesterol lowering agents
- HMG-CoA reductase inhibitors lovastatin, simvastatin and pravastatin, fluvastatin.
- atorvastatin and other statins
- sequestrants cholesterolestyramine, colestipol and a dialkylaminoalkyl derivatives of a cross-linked dextran
- nicotinyl alcohol nicotinic acid or a salt thereof e.g., proliferator-activater receptor ⁇ agonists such as fenofibric acid derivatives (gemfibrozil, clofibrat, fenofibrate and benzafibrate).
- inhibitors of cholesterol absorption for example beta-sitosterol and (acyl CoA holesterol acyltransferase) inhibitors for example melinamide,
- probucol. vitamin E, and (vii) thyromimetics
- PPAR ⁇ agonists such as those disclosed in W097/28149;
- antiobesity compounds such as fenfluramine, dexfenfluramine, phentermine, sibutramine, orlistat, or ⁇ 3 adrenergic receptor agonists;
- feeding behavior modifying agents such as neuropeptide Y antagonists (e.g. neuropeptide Y5) such as those disclosed in WO 97/19682, WO 97/20820, WO 97 ⁇ '20821, WO 97/20822 and WO 97/20823;
- PPAR ⁇ agonists such as described in WO 97'36579 by Glaxo;
- growth hormone secretagogues such as MK-0677.
- agents useful in the treatment of male and/or female sexual dysfunction such as phosphodiester V inhibitors such as sildenafil, and ⁇ -2 adrenergic receptor antagonists.
- A. Binding Assay The membrane binding assay is used to identify competitive inhibitors of 125 I- ⁇ -NDP-MSH binding to cloned human MCRs expressed in L- or CHO- cells.
- Cell lines expressing melanocortin receptors are grown in T-l 80 flasks containing selective medium of the composiiton: 1 L Dulbecco's modified Eagles Medium (DMEM) with 4.5 g L-glucose, 25 mM Hepes, without sodium pyruvate, (Gibco/BRl); 100 ml 10% heat-inactivated fetal bovine serum (Sigma); 10 ml 10,000 unit/ml penicillin & 10,000 ug/ml streptomycin (Gibco/BRl); 10 ml 200 mM L-glutamine (Gibco BRl); 1 mg/ml Geneticin (G418) (Gibco/BRl). The cells are grown at 37°C with CO2 and humidity control until the
- the medium is poured off and 10 mls/monolayer of enzyme-free dissociation media (Specialty Media Inc.) is added.
- the cells are incubated at 37°C for 10 minutes or until cells slough off when flask is banged against hand.
- the cells are harvested into 200 ml centrifuge tubes and spun at 1000 rpm, 4° C, for 10 min. The supernatant is discarded and the cells are resuspended in 5 mls/monolayer membrane preparation buffer having the composition: 10 mM Tris pH 7.2-7.4; 4 ug/ml Leupeptin (Sigma); 10 uM Phosphoramidon (Boehringer Mannheim); 40 ug/ml Bacitracin (Sigma); 5 ug/ml Aprotinin (Sigma); 10 mM Pefabloc (Boehringer Mannheim). The cells are homogenized with motor-driven dounce (Talboy setting 40), using 10 strokes and the homogenate centrifuged at 6,000 rpm, 4 C, for 15 minutes.
- pellets are resuspended in 0.2 mls/monolayer membrane prep buffer and aliquots are placed in tubes (500-1000 ul tube) and quick frozen in liquid nitrogen and then store at -80 ° C.
- Test compounds or unlabelled NDP- ⁇ -MSH is added to 100 ⁇ L of membrane binding buffer to a final concentration of 1 ⁇ M.
- the membrane binding buffer has the composition: 50 mM Tris pH 7.2; 2 mM CaC12; 1 mM MgC12; 5 mM KCl; 0.2% BSA; 4 ug/ml Leupeptin (SIGMA); 10 uM Phosphoramidon (Boehringer Mannheim); 40 ug ml Bacitracin (SIGMA); 5 ug ml Aprotinin (SIGMA); and 10 mM Pefabloc (Boehringer Mannheim).
- membrane binding buffer containing 10-40 ug membrane protein is added, followed by 100 ⁇ M 125I-NDP- ⁇ - MSH to final concentration of 100 pM.
- the resulting mixture is vortexed briefly and incubated for 90-120 min at room temp while shaking.
- the mixture is filtered with Packard Microplate 196 filter apparatus using Packard Unifilter 96-well GF/C filter with 0.1% polyethyleneimine (Sigma).
- the filter is washed (5 times with a total of 10 ml per well) with room temperature of filter wash having the composition: 50mM Tris-HCl pH 7.2 and 20 mM NaCl.
- the filter is dried, and the bottom sealed and 50 ul of Packard Microscint-20 is added to each well. The top is sealed and the radioactivity quantitated in a Packard Topcount Microplate Scintillation counter.
- Functional assay Functional cell based assays are developed to discriminate agonists and antagonists.
- Cells for example, CHO- or L-cells or other eukaryotic cells
- a human melanocortin receptor see e.g. Yang-YK; Ollmann-MM; Wilson-BD; Dickinson-C; Yamada-T; Barsh-GS; Gantz-I; Mol-Endocrinol. 1997 Mar; 11(3): 274-80
- Ca and Mg free phosphate buffered saline 14190-136, Life Technologies, Gaithersburg, MD
- enzyme free dissociation buffer S-014-B, Specialty Media, Lavellette, NJ
- cAMP detection assay RPA556
- the amount of cAMP production which results from an unknown compound is compared to that amount of cAMP produced in response to alpha-MSH which is defined as a 100 % agonist.
- the EC50 is defined as the compound concentration which results in half maximal stimulation, when compared to its own maxim level of stimulation.
- Antagonist activity is defined as the ability of a compound to block cAMP production in response to alpha-MSH.
- Solution of test compounds and suspension of receptor containing cells are prepared and mixed as described above; the mixture is incubated for 15 min., and an EC50 dose (approximately 10 nM alpha-MSH) is added to the cells.
- the assay is terminated at 45 min. and cAMP quantitated as above. Percent inhibition is determined by comparing the amount of cAMP produced in the presence to that produced in the absence of test compound.
- the lower torso and hind limbs are restrained with a non- adhesive material (vetrap).
- Penile responses will be observed, typically termed ex copula genital reflex tests. Typically, a series of penile erections will occur spontaneously within a few minutes after sheath retraction.
- the types of normal reflexogenic erectile responses include elongation, engorgement, cup and flip. An elongation is classified as an extension of the penile body. Engorgement is a dilation of the glans penis.
- a cup is defined as an intense erection where the distal margin of the glans penis momentarily flares open to form a cup.
- a flip is a dorsiflexion of the penile body.
- Baseline and or vehicle evaluations are conducted to determine how and if an animal will respond. Some animals have a long duration until the first response while others are non-responders altogether. During this baseline evaluation latency to first response, number and type of responses are recorded. The testing time frame is 15 minutes after the first response. After a minimum of 1 day between evaluations, these same animals are administered the test compound at 20 mg/kg and evaluated for penile reflexes. All evaluations are videotaped and scored later. Data are collected and analyzed using paired 2 tailed t-tests to compared baseline and/ or vehicle evaluations to drug treated evaluations for individual animals. Groups of a minimum of 4 animals are utilized to reduce variability.
- mice can be dosed by a number of routes of administration depending on the nature of the study to be performed.
- the routes of administration includes intravenous (IV), intraperitoneal (IP), subcutaneous (SC) and intracerebral ventricular (ICV).
- E. Models of Female sexual Dysfunctioin Rodent assays relevant to female sexual receptivity include the behavioral model of lordosis and direct observations of copulatory activity. There is also a urethrogenital reflex model in anesthetized spinally transected rats for measuring orgasm in both male and female rats. These and other established animal models of female sexual dysfunction are described in McKenna KE et al, A Model For The Study Of Sexual Function In Anesthetized Male And Female Rats, Am. J. Physiol. (Regulatory Integrative Comp.
- the preparation of compounds of Formula I of the present invention may be carried out in sequential or convergent synthetic routes. Syntheses detailing the preparation of the compounds of Formula I in a sequential manner are presented in the following reaction schemes.
- the instant compounds are generally isolated in the form of their pharmaceutically acceptable salts, such as those described previously hereinabove.
- standard peptide coupling reaction conditions is used repeatedly here, and it means coupling a carboxylic acid with an amine using an acid activating agent such as EDC, DCC, and BOP in a inert solvent such as dichloromethane in the presence of a catalyst such as HOBT.
- an acid activating agent such as EDC, DCC, and BOP
- a inert solvent such as dichloromethane
- HOBT a catalyst
- protective groups for amine and carboxylic acid to facilitate the desired reaction and minimize undesired reactions are well documented. Conditions required to remove protecting groups which may be present and can be found in Greene, T, and Wuts, P. G. M., Protective Groups in Organic Synthesis, John Wiley & Sons, Inc.. New York, NY 1991. CBZ and BOC are used extensively in the synthesis, and their removal conditions are known to those skilled in the art.
- removal of CBZ groups can be achieved by a number of methods known in the art; for example, catalytic hydrogenation with hydrogen in the presence of a noble metal or its oxide such as palladium on activated carbon in a pro tic solvent such as ethanol.
- removal of CBZ groups can also be achieved by treatment with a solution of hydrogen bromide in acetic acid, or by treatment with a mixture of TFA and dimethylsulfide.
- Removal of BOC protecting groups is carried out in a solvent such as methylene chloride or methanol or ethyl acetate, with a strong acid, such as trifluoroacetic acid or hydrochloric acid or hydrogen chloride gas.
- the protected amino acid derivatives 1 are, in many cases, commercially available, where the protecting group L is, for example, BOC or CBZ groups.
- Other protected amino acid derivatives 1 can be prepared by literature methods (Williams, R. M. Synthesis of Optically Active a-Amino Acids, Pergamon Press: Oxford, 1989).
- Many of the piperidines of Formula 2 are either commercially available or known in the literature and others can be prepared following literature methods described for analogous compounds. Some of these methods are illustrated in the subsequent schemes. The skills required in carrying out the reaction and purification of the resulting reaction products are known to those in the art. Purification procedures include crystallization, normal phase or reverse phase chromatography.
- amino acid ester intermediates of Formula 5 wherein L' is a small alkyl such as methyl or ethyl or a benzyl or allyl unit, can be synthesized by well documented methods in the literature. Coupling of intermediates 4 and 5 under standard peptide coupling conditions followed by removal of the ester group L' yields the intermediate 6. Compounds of formula I are obtained by coupling intermediates of Formula 6 to spiropiperidines of formula 2 under standard peptide coupling reaction conditions, followed by the removal of the amino protecting group, L.
- the compounds of the present invention may also be prepared from a variety of substituted natural and unnatural amino acids of formulas 8.
- the spiropiperidines of formula 12 may be prepared by a number of methods, including the syntheses described below. In cases where a sulfide is present in the molecule, it may be oxidized to a sulfoxide or to a sulfone with oxidizing agents such as sodium periodate, m-chloroperbenzoic acid or Oxone " in an solvent such as dichloromethane, alcohol or water or their mixtures.
- oxidizing agents such as sodium periodate, m-chloroperbenzoic acid or Oxone
- the indoline nitrogen of 13 can be reacted by with a variety of electrophiles to yield spiro piperidines of formula 14, wherein the substitutent can be a variety of functionalities, including Rb, SO2R a , SO2N(Rb)2, CORa, CON(Rb) 2 .
- Compound 13 can be reacted with, for example, isocyanates in an inert solvent like dichloromethane to yield urea derivatives, chloroformates in an inert solvent such as dichloromethane to yield carbamates, acid chlorides, anhydrides, or acyl imidazoles to generate amides, sulfonyl chlorides to generate sulfonamides. sulfamyl chlorides to yield sulfamides.
- the indoline nitrogen of 13 can be reductively alkylated with aldehydes with conditions known in the art.
- Aromatic units, including substituted heteroaryl groups, can be introduced by reacting 13 with a fluoro phenyl or fluoro heteroaryl reagent. This chemistry is detailed by H. Ong et al J. Med. Chem. 1983, 23, 981-986.
- Demethylation of 14 be accomplished by reacting it with cyanogen bromide and potassium carbonate in an inert solvent solvent such as dichloromethane to yield a cyanamide which can reduced to give 15 by treatment with lithium aluminum hydride in refluxing tetrahydrofuran, refluxing strong acid like aqueous hydrochloric acid, or with Grignard reagents like methyl magnesium bromide.
- demethylation of 14 can be effected with the ACE-C1 method as described in R. Olofson et al. J Org.
- Debenzylation can be accomplished by reductive methods including hydrogenation in the presence of platinum or palladium catalyst in a protic solvent like methanol.
- debenzylation of 14 can be effected with the ACE-C1 method as described in R. Olofson et al. J. Org. Chem. 1984, 49, 2795 and references therein.
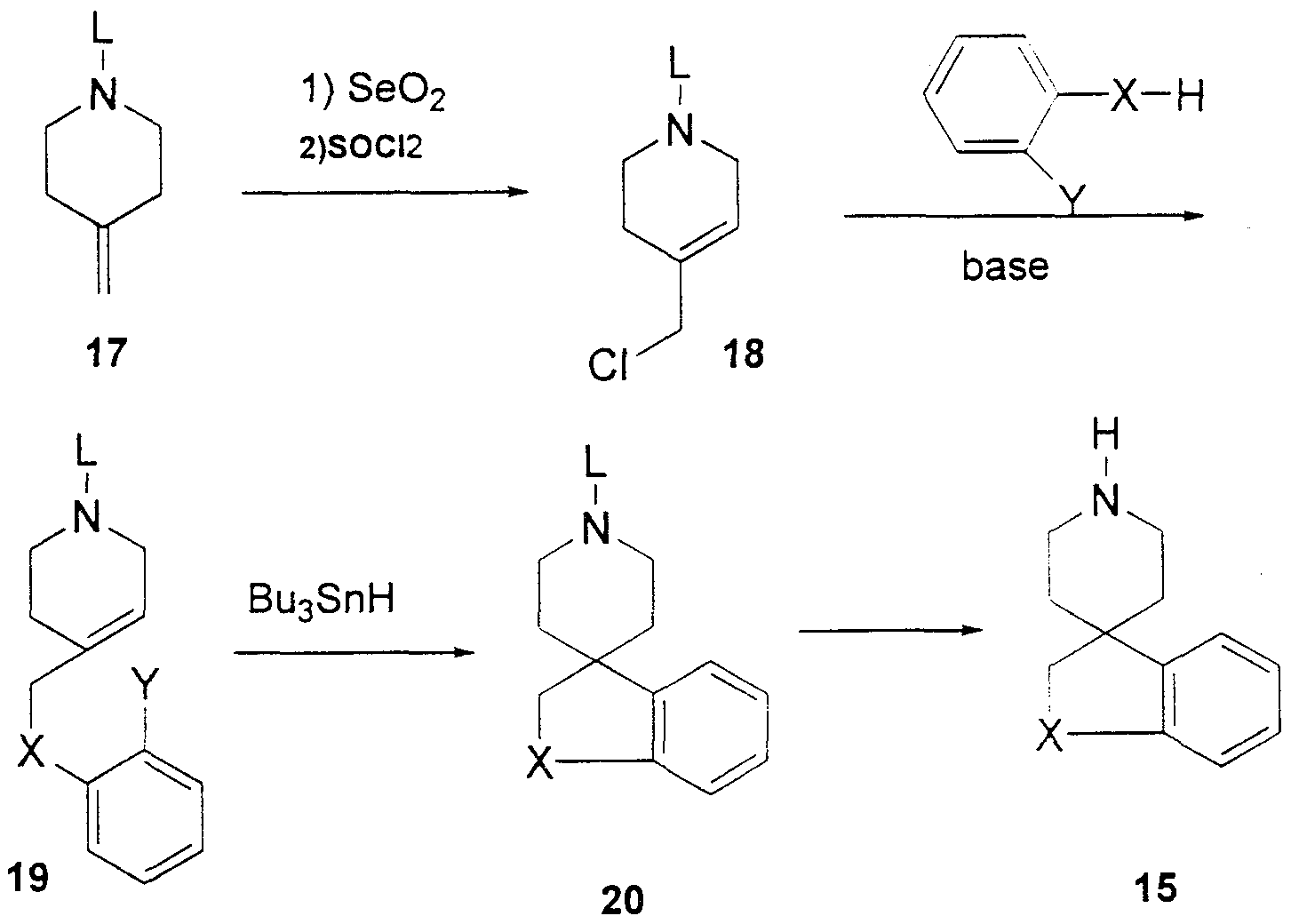
- the spiro heterocyclic compounds of formula 15 can be prepared by a number of methods, including the syntheses as described in Scheme 7. Allylic oxidation of the protected piperidine 17 is accomplished by classical methods familiar to those skilled in the art (Rabjohn, N. Org. React. 1976, 24, 261). The resulting allylic alcohol is treated with thionyl chloride in an inert solvent such as benzene to provide the corresponding chloride 18.
- X S(O)
- SO2 sulfone
- sodium periodate is often used for the synthesis of sulfoxides
- Oxone is used for the synthesis of sulfones. Removal of the protecting group provides the amine 16 which then can be elaborated to melanocortin agonists.
- spiro indanone 21 provides easy access to spiroindanyl intermediates containing acid and ester groups.
- This chemistry is described in Scheme 10.
- Treatment of 21 with a base in an inert solvent such as THF followed by the addition of a triflating agent provides the enol triflate.
- Carboxylation of the enol triflate according to the procedure of Cacchi, S. Tetrahedron Letters, 1985, 1109-1112 provides the ester 23.
- Hydrogenation of 23 using a palladium catalyst in an inert solvent provides the saturated ester 24.
- the protecting group can then be removed as described above and the resulting amine can be incorporated into the subject compound via the chemistry depicted in earlier schemes.
- estonification of the ester of 24 provides an acid which can be conveniently derivatized as for example reaction with an amine in the presence of a coupling agent such as EDC gives amides, which can then be incorporated into final compounds.
- the ester 24 may also be reduced to a primary alcohol with LAH and to a aldehyde with DIBALH. Reductive alkylation of the aldehyde with ammonium acetate and sodium cyanoborohydride affords an amino methyl analog. These aminomethyl analogs may then be further reacted with acylating and sulfonylating agents to afford additional melanocortin compounds of the general formula I.
- spiroindanes can be hydrogenated with Pt/C or Rh alumina as catalysts in solvents such as methanol. ethanol or acetic acid to afford corresponding perhydroindanes. High pressures are often required to carry out this saturation reaction.
- the L protecting group can be removed by standard methods as discussed above.
- Chiral acids are available by a variety of methods known to those skilled in the art including asymmetric catalytic hydrogenation and resolution of a pair of diastereomeric salts formed by reaction with a chiral amine such as D or L ⁇ - methylbenzylamine.
- the absolute stereochemistry can be determined in a number of ways including X-ray crystallography of a suitable crystalline derivative.
- a substituted phenyl alanine derivative 25 can be treated with aqueous formaldehyde in concentrated hydrochloric acid to afford, after protection of the amino functionality in a second step by well documented methods, the tetrahydroisoquinoline compound 26.
- This reaction can also be effected with heterocyclic amino acids such as 2- and 3-thienyl Ala. Since the above chemistry works generally with retention of stereochemistry, D- and L-amino acids of general formula 5 can be prepared from D- and L- amino acids.
- a strong base such as NaH in DMF
- esters of formula 28 with alkali leads to formation of the corresponding mono carboxylic acid which can be treated with refluxing hydrochloric to affect hydrolysis of the acetamide derivative to provide amino acid of formula 29.
- standard protection of the amino functionality provides intermediates of formula 30.
- Saturated amino side chains of formula 31 can be prepared by hydrogenating compounds of formula 30 in the presence of rhodium or platinum catalysts.
- compound 30 can be hydrogenated in the presence of 5% Rh on alumina to give compound 31.
- Individual diastereomers of 31 can be resolved via classical resolution methods.
- Schemes 14 illustrates one method for the preparation of tetrahydroisoquinolineacetic acid of formula 33. This is carried out conveniently by the Arndt-Eistert reaction which proceeds with retention of stereochemistry. Other methods involve require reduction of the acid or its ester derivative to an alcohol, conversion of the alcohol to a leaving group such as a mesylate or halide, displacement of it with cyanide anion and hydrolysis of the nitrile to the carboxylic acid by well documented literature methods.
- the starting material, l,2-dihydro-5-fluoro-l-methanesulfonylspiro[3H-indole-3,4'- piperidine] hydrochloride may be prepared according to general method disclosed in US Patent 5536716.
- Step A Preparation of 3 -oxo- 1 '-(t-butyloxycarbony l)spiro [ 1 H-indan- 1.4'-piperidine]
- Step B Preparation of 3-[(trifluoromethanesulfonyl)oxy]-l'-(t- butyloxycarbonyl)spiro[lH-indene-l ,4'-piperidine]
- Step C Preparation of 3-(ethoxycarbonyl)-l'-(t-butyloxycarbonyl
- Step D Preparation of 3-(carboxy)-l'-(t-butyloxycarbonyl)spiro[lH-indan-l, 4'- piperidine]
- Step E Preparation of 3 S-3-(carboxy)- 1 '-(t-butyloxycarbonyl)spiro [ 1 H-indan- 1 ,4'- piperidine]
- Step F Preparation of 3R-3-(carboxy)-r-(t-butyloxycarbonyl)spiro[lH-indan-l,4'- piperidine]
- Step G Preparation of 3R-3-[[(benzyloxy)carbonyl]amino]- -(t- buty loxy carbony l)spiro [ 1 H-indan- 1 ,4'-piperidine]
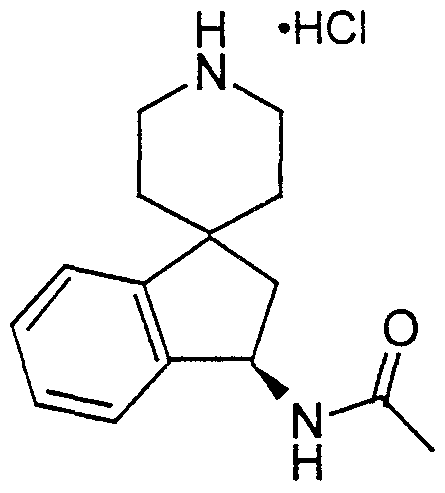
- Step H Preparation of 3R-3-amino-l'-(t-butyloxycarbonyl)spiro[lH-indan-l,4'- piperidine] hydrochloride salt
- Step G of Intermediate 10 The general procedure described in Step G of Intermediate 10 was followed using cvclopropylamine instead of benzyl alchol to react with the isocyanato compound to provide the N-Boc protected title compound.
- the N-Boc protecting group was removed according to the general procedure described in Intermediate 11 to provide the title compound.
- Step A Preparation of 3S-N-Boc-3-decahydroisoquinolinecarboxylic acid
- N-Boc-(I)-Tic methyl ester (1.17 g) as a yellow oil.
- a solution of N-Boc-(I)-Tic methyl ester (1.05 g, 3.60 mmol) and 12 mL of methanol was charged with 5% rhodium on alumina (0.53 g) and then heated at 55-60 °C under 40 psi of hydrogen for 36 h.
- Step A Preparation of [3S(3 ⁇ ,4a ⁇ ,8a ⁇ )]-N-Boc-decahydro-3-isoquinolinecarboxylic acid
Abstract
Description






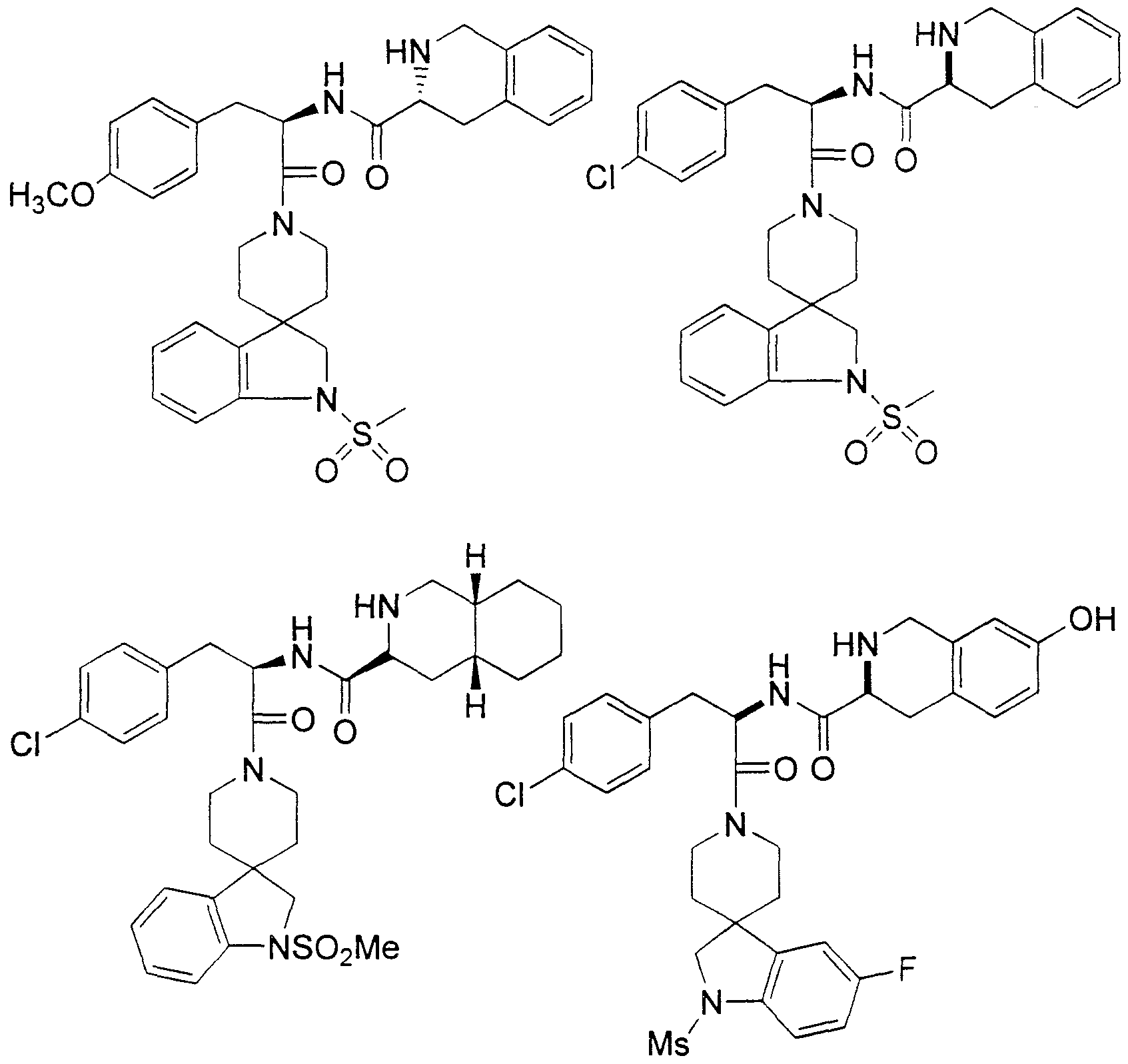























































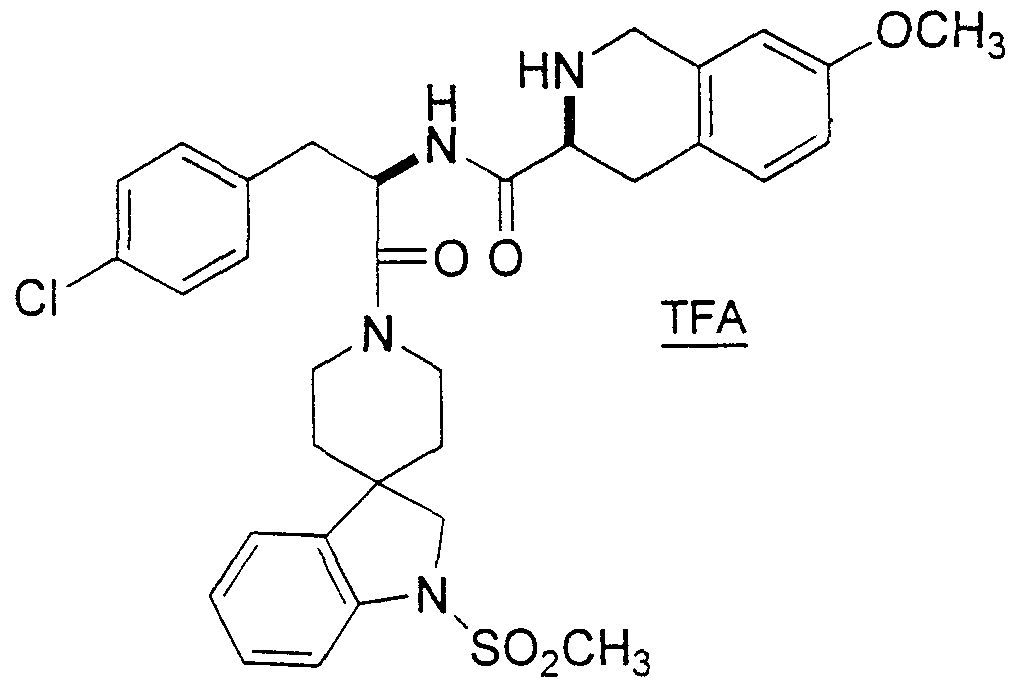




Claims












Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002334551A CA2334551A1 (en) | 1998-06-11 | 1999-06-10 | Spiropiperidine derivatives as melanocortin receptor agonists |
| EP99930220A EP1085869A4 (en) | 1998-06-11 | 1999-06-10 | Spiropiperidine derivatives as melanocortin receptor agonists |
| JP2000553071A JP2002517444A (en) | 1998-06-11 | 1999-06-10 | Spiropiperidine derivatives as melanocortin receptor agonists |
| AU46801/99A AU742425B2 (en) | 1998-06-11 | 1999-06-10 | Spiropiperidine derivatives as melanocortin receptor agonists |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US8890898P | 1998-06-11 | 1998-06-11 | |
| US60/088,908 | 1998-06-11 | ||
| GBGB9817179.6A GB9817179D0 (en) | 1998-08-06 | 1998-08-06 | Spiropiperidine derivatives as melanocortin receptor agonists |
| GB9817179.6 | 1998-08-06 | ||
| US12326099P | 1999-03-08 | 1999-03-08 | |
| US60/123,260 | 1999-03-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999064002A1 true WO1999064002A1 (en) | 1999-12-16 |
Family
ID=27269429
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1999/013252 WO1999064002A1 (en) | 1998-06-11 | 1999-06-10 | Spiropiperidine derivatives as melanocortin receptor agonists |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1085869A4 (en) |
| JP (1) | JP2002517444A (en) |
| AU (1) | AU742425B2 (en) |
| CA (1) | CA2334551A1 (en) |
| WO (1) | WO1999064002A1 (en) |
Cited By (146)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001000224A1 (en) * | 1999-06-29 | 2001-01-04 | Palatin Technologies Inc. | Compositions and methods for treatment of sexual dysfunction |
| WO2001010842A2 (en) * | 1999-08-04 | 2001-02-15 | Millennium Pharmaceuticals, Inc. | Melanocortin-4 receptor binding compounds and methods of use thereof |
| WO2001055109A1 (en) * | 2000-01-28 | 2001-08-02 | Melacure Therapeutics Ab | Aromatic amides acting on melanocortin receptors |
| WO2001055107A2 (en) * | 2000-01-28 | 2001-08-02 | Melacure Therapeutics Ab | Aromatic amines and amides acting on the melanocortin receptors |
| WO2001074844A2 (en) * | 2000-04-04 | 2001-10-11 | F. Hoffmann-La Roche Ag | Selective linear peptides with melanocortin-4 receptor (mc4-r) agonist activity |
| WO2002000654A1 (en) * | 2000-06-28 | 2002-01-03 | Pfizer Products Inc. | Melanocortin receptor ligands |
| WO2002012178A1 (en) * | 2000-08-07 | 2002-02-14 | Melacure Therapeutics Ab | Compounds acting as melanocortin receptor ligands |
| US6350760B1 (en) | 1999-06-04 | 2002-02-26 | Merck & Co., Inc. | Substituted piperidines as melanocortin-4 receptor agonists |
| WO2002018327A2 (en) * | 2000-08-31 | 2002-03-07 | Chiron Corporation | Guanidinobenzamides as mc4-r agonists |
| WO2002059095A1 (en) * | 2001-01-23 | 2002-08-01 | Eli Lilly And Company | Melanocortin receptor agonists |
| WO2002059107A1 (en) * | 2001-01-23 | 2002-08-01 | Eli Lilly And Company | Substituted piperidines/piperazines as melanocortin receptor agonists |
| WO2002059108A1 (en) * | 2001-01-23 | 2002-08-01 | Eli Lilly And Company | Melanocortin receptor agonists |
| WO2002062766A2 (en) * | 2001-02-07 | 2002-08-15 | Millennium Pharmaceuticals, Inc. | Melanocortin-4 receptor binding compounds and methods of use thereof |
| US6458790B2 (en) | 2000-03-23 | 2002-10-01 | Merck & Co., Inc. | Substituted piperidines as melanocortin receptor agonists |
| US6472398B1 (en) | 2000-03-23 | 2002-10-29 | Merck & Co., Inc. | Spiropiperidine derivatives as melanocortin receptor agonists |
| WO2002094825A1 (en) * | 2001-05-22 | 2002-11-28 | Banyu Pharmaceutical Co., Ltd. | Novel spiropiperidine derivative |
| WO2003009850A1 (en) * | 2001-07-25 | 2003-02-06 | Amgen Inc. | Substituted piperazines as modulators of the melanocortin receptor |
| EP1285658A2 (en) * | 2001-08-21 | 2003-02-26 | Pfizer Productors Inc. | Treatments for female sexual dysfunction |
| EP1289526A1 (en) * | 2000-05-30 | 2003-03-12 | Merck & Co., Inc. | Melanocortin receptor agonists |
| EP1295608A1 (en) * | 2000-06-27 | 2003-03-26 | Taisho Pharmaceutical Co., Ltd | Remedial agent for anxiety neurosis or depression and piperazine derivative |
| KR20030027439A (en) * | 2001-09-28 | 2003-04-07 | 주식회사 엘지생명과학 | Melanocortin receptor agonists |
| KR20030035589A (en) * | 2001-10-31 | 2003-05-09 | 주식회사 엘지생명과학 | Melanocortin receptor agonists |
| KR20030035592A (en) * | 2001-10-31 | 2003-05-09 | 주식회사 엘지생명과학 | Melanocortin receptor agonists |
| WO2003061660A1 (en) * | 2002-01-23 | 2003-07-31 | Eli Lilly And Company | Melanocortin receptor agonists |
| WO2003066587A2 (en) * | 2002-02-04 | 2003-08-14 | Chiron Corporation | Piperidine or pyrrolidine derivatives having an aminoacyl substituted side chain as melancortin-4 receptor agonists |
| WO2003094918A1 (en) * | 2002-05-10 | 2003-11-20 | Neurocrine Biosciences, Inc. | Substituted piperazine as melanocortin receptors ligands |
| EP1363898A1 (en) * | 2001-03-02 | 2003-11-26 | Bristol-Myers Squibb Company | Compounds useful as modulators of melanocortin receptors and pharmaceutical compositions comprising same |
| WO2004002986A2 (en) | 2002-06-28 | 2004-01-08 | Banyu Pharmaceutical Co., Ltd. | Novel benzimidazole derivatives |
| US6699873B1 (en) | 1999-08-04 | 2004-03-02 | Millennium Pharmaceuticals, Inc. | Melanocortin-4 receptor binding compounds and methods of use thereof |
| US6716840B2 (en) | 2001-04-09 | 2004-04-06 | Chiron Corporation | Guanidino compounds |
| EP1425029A1 (en) * | 2001-08-10 | 2004-06-09 | Palatin Technologies, Inc. | Peptidomimetics of biologically active metallopeptides |
| US6767915B2 (en) | 2000-08-23 | 2004-07-27 | Merck & Co., Inc. | Substituted piperidines as melanocortin receptor agonists |
| WO2004083209A1 (en) * | 2003-03-20 | 2004-09-30 | Santhera Pharmaceuticals (Schweiz) Gmbh | Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators |
| US6818658B2 (en) | 2001-02-28 | 2004-11-16 | Merck & Co., Inc. | Acylated piperidine derivatives as melanocortin-4 receptor agonists |
| WO2005028438A1 (en) | 2003-09-22 | 2005-03-31 | Banyu Pharmaceutical Co., Ltd. | Novel piperidine derivative |
| US6911447B2 (en) | 2001-04-25 | 2005-06-28 | The Procter & Gamble Company | Melanocortin receptor ligands |
| WO2005058836A1 (en) * | 2003-12-12 | 2005-06-30 | Syngenta Participations Ag | Insecticidal spiroindane derivatives |
| US6916812B2 (en) | 2001-10-09 | 2005-07-12 | Bristol-Myers Squibb Company | Alpha-aminoamide derivatives as melanocortin agonists |
| WO2005097759A1 (en) | 2004-03-29 | 2005-10-20 | Merck & Co., Inc. | Diaryltriazoles as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2006017542A1 (en) | 2004-08-06 | 2006-02-16 | Merck & Co., Inc. | Sulfonyl compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| US7012084B2 (en) | 2001-02-28 | 2006-03-14 | Merck & Co., Inc. | Acylated piperidine derivatives as melanocortin-4 receptor agonists |
| US7026335B2 (en) | 2002-04-30 | 2006-04-11 | The Procter & Gamble Co. | Melanocortin receptor ligands |
| US7034033B2 (en) | 2002-05-23 | 2006-04-25 | Chiron Corporation | Substituted quinazolinone compounds |
| US7045527B2 (en) | 2002-09-24 | 2006-05-16 | Amgen Inc. | Piperidine derivatives |
| US7049323B2 (en) | 2003-04-25 | 2006-05-23 | Bristol-Myers Squibb Company | Amidoheterocycles as modulators of the melanocortin-4 receptor |
| US7049331B2 (en) | 2001-11-08 | 2006-05-23 | Ortho-Mcneil Pharmaceutical, Inc. | 1,2,4-thiadiazole derivatives as melanocortin receptor modulators |
| US7115628B2 (en) | 2001-07-18 | 2006-10-03 | Merck & Co., Inc. | Bridged piperidine derivatives as melanocortin receptor agonists |
| US7125885B2 (en) | 2001-05-04 | 2006-10-24 | Amgen Inc. | Fused heterocyclic compounds |
| US7132539B2 (en) | 2002-10-23 | 2006-11-07 | The Procter & Gamble Company | Melanocortin receptor ligands |
| WO2006129826A1 (en) | 2005-05-30 | 2006-12-07 | Banyu Pharmaceutical Co., Ltd. | Novel piperidine derivative |
| US7160886B2 (en) | 2003-03-03 | 2007-01-09 | Merck & Co., Inc. | Acylated piperazine derivatives as melanocortin-4 receptor agonists |
| US7176279B2 (en) | 2000-06-28 | 2007-02-13 | Palatin Technologies, Inc. | Cyclic peptide compositions and methods for treatment of sexual dysfunction |
| WO2007018248A1 (en) | 2005-08-10 | 2007-02-15 | Banyu Pharmaceutical Co., Ltd. | Pyridone compound |
| WO2007024004A1 (en) | 2005-08-24 | 2007-03-01 | Banyu Pharmaceutical Co., Ltd. | Phenylpyridone derivative |
| WO2007029847A1 (en) | 2005-09-07 | 2007-03-15 | Banyu Pharmaceutical Co., Ltd. | Bicyclic aromatic substituted pyridone derivative |
| WO2007041052A2 (en) | 2005-09-29 | 2007-04-12 | Merck & Co., Inc. | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| WO2007049798A1 (en) | 2005-10-27 | 2007-05-03 | Banyu Pharmaceutical Co., Ltd. | Novel benzoxathiin derivative |
| WO2007055418A1 (en) | 2005-11-10 | 2007-05-18 | Banyu Pharmaceutical Co., Ltd. | Aza-substituted spiro derivative |
| EP1801098A1 (en) | 2005-12-16 | 2007-06-27 | Merck Sante | 2-Adamantylurea derivatives as selective 11B-HSD1 inhibitors |
| US7253179B2 (en) | 2002-11-06 | 2007-08-07 | Amgen Inc. | Fused heterocyclic compounds |
| US7276520B2 (en) | 2003-03-26 | 2007-10-02 | Merck & Co., Inc. | Bicyclic piperidine derivatives as melanocortin-4 receptor agonists |
| US7307063B2 (en) | 2001-02-13 | 2007-12-11 | Palatin Technologies, Inc. | Melanocortin metallopeptides for treatment of sexual dysfunction |
| US7319107B2 (en) | 2001-11-08 | 2008-01-15 | Johnson & Johnson Consumer Companies, Inc. | 1,2,4-thiadiazolium derivatives as melanocortin receptor modulators |
| EP1880996A1 (en) | 2002-06-14 | 2008-01-23 | Syngenta Limited | Spiroindolinepiperidine derivatives |
| US7323462B2 (en) | 2002-12-10 | 2008-01-29 | Pfizer Inc. | Morpholine dopamine agonists |
| US7329673B2 (en) | 2003-04-04 | 2008-02-12 | Merck & Co., Inc. | Acylated spiropiperidine derivatives as melanocortin-4 receptor agonists |
| WO2007100664A3 (en) * | 2006-02-22 | 2008-03-06 | Vertex Pharma | Modulators of muscarinic receptors |
| WO2008038692A1 (en) | 2006-09-28 | 2008-04-03 | Banyu Pharmaceutical Co., Ltd. | Diaryl ketimine derivative |
| WO2008039863A2 (en) * | 2006-09-27 | 2008-04-03 | Braincells, Inc. | Composition comprising a melanocortin receptor (mcr) modulating agent alone or in combination with a second neurogenic agent for treating nervous system disorders |
| WO2008039418A2 (en) * | 2006-09-27 | 2008-04-03 | Merck & Co., Inc. | Acylated piperidine derivatives as melanocortin-4 receptor modulators |
| US7368453B2 (en) | 2003-11-19 | 2008-05-06 | Chiron Corporation | Quinazolinone compounds with reduced bioaccumulation |
| US7375125B2 (en) | 1999-08-04 | 2008-05-20 | Ore Pharmaceuticals, Inc. | Melanocortin-4 receptor binding compounds and methods of use thereof |
| WO2008071980A1 (en) | 2006-12-14 | 2008-06-19 | Acure Pharma Ab | Novel aminoguanidines as melanocortin receptor ligands. |
| WO2008074384A1 (en) | 2006-12-21 | 2008-06-26 | Merck Patent Gmbh | 2-ADAMANTYL-BUTYRAMIDE DERIVATIVES AS SELECTIVE 11βETA-HSD1 INHIBITORS |
| US7414057B2 (en) | 2002-09-11 | 2008-08-19 | Merck & Co., Inc. | Piperazine urea derivatives as melanocortin-4 receptor agonists |
| US7420059B2 (en) | 2003-11-20 | 2008-09-02 | Bristol-Myers Squibb Company | HMG-CoA reductase inhibitors and method |
| WO2008120653A1 (en) | 2007-04-02 | 2008-10-09 | Banyu Pharmaceutical Co., Ltd. | Indoledione derivative |
| AU2004202804B2 (en) * | 1999-08-04 | 2009-01-29 | Ore Pharmaceuticals Inc. | Melanocortin-4 Receptor Binding Compounds and Methods of use thereof |
| EP2088154A1 (en) | 2004-03-09 | 2009-08-12 | Ironwood Pharmaceuticals, Inc. | Methods and compositions for the treatment of gastrointestinal disorders |
| WO2009110510A1 (en) | 2008-03-06 | 2009-09-11 | 萬有製薬株式会社 | Alkylaminopyridine derivative |
| WO2009119726A1 (en) | 2008-03-28 | 2009-10-01 | 萬有製薬株式会社 | Diarylmethylamide derivative having antagonistic activity on melanin-concentrating hormone receptor |
| EP2110374A1 (en) | 2008-04-18 | 2009-10-21 | Merck Sante | Benzofurane, benzothiophene, benzothiazol derivatives as FXR modulators |
| US7618987B2 (en) | 2004-07-19 | 2009-11-17 | Merck & Co., Inc. | Acylated piperidine derivatives as melanocortin 4-receptor agonists |
| US7625909B2 (en) | 2003-05-23 | 2009-12-01 | Novartis Vaccines And Diagnostics, Inc. | Substituted quinazolinone compounds |
| WO2009154132A1 (en) | 2008-06-19 | 2009-12-23 | 萬有製薬株式会社 | Spirodiamine-diarylketoxime derivative |
| US7652024B2 (en) | 2005-10-18 | 2010-01-26 | Merck Sharp & Dohme Corp. | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| WO2010013595A1 (en) | 2008-07-30 | 2010-02-04 | 萬有製薬株式会社 | (5-membered)-(5-membered) or (5-membered)-(6-membered) fused ring cycloalkylamine derivative |
| US7696201B2 (en) | 2006-08-15 | 2010-04-13 | Vertex Pharmaceuticals Incorporated | Modulators of muscarinic receptors |
| WO2010047982A1 (en) | 2008-10-22 | 2010-04-29 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2010051206A1 (en) | 2008-10-31 | 2010-05-06 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2010056717A1 (en) | 2008-11-17 | 2010-05-20 | Merck Sharp & Dohme Corp. | Substituted bicyclic amines for the treatment of diabetes |
| US7795378B2 (en) | 2002-07-09 | 2010-09-14 | Palatin Technologies, Inc. | Peptide compositions for treatment of sexual dysfunction |
| US7858790B2 (en) | 2006-06-29 | 2010-12-28 | Vertex Pharmaceuticals Incorporated | Modulators of muscarinic receptors |
| US7858635B2 (en) | 2005-12-22 | 2010-12-28 | Vertex Pharmaceuticals Incorporated | Spiro compounds as modulators of muscarinic receptors |
| WO2011011508A1 (en) | 2009-07-23 | 2011-01-27 | Schering Corporation | Benzo-fused oxazepine compounds as stearoyl-coenzyme a delta-9 desaturase inhibitors |
| WO2011011506A1 (en) | 2009-07-23 | 2011-01-27 | Schering Corporation | Spirocyclic oxazepine compounds as stearoyl-coenzyme a delta-9 desaturase inhibitors |
| EP2305352A1 (en) | 2004-04-02 | 2011-04-06 | Merck Sharp & Dohme Corp. | 5-alpha-reductase inhibitors for use in the treatment of men with metabolic and anthropometric disorders |
| WO2011058193A1 (en) | 2009-11-16 | 2011-05-19 | Mellitech | [1,5]-diazocin derivatives |
| WO2011069038A2 (en) | 2009-12-03 | 2011-06-09 | Synergy Pharmaceuticals, Inc. | Agonists of guanylate cyclase useful for the treatment of hypercholesterolemia, atherosclerosis, coronary heart disease, gallstone, obesity and other cardiovascular diseases |
| EP2332526A2 (en) | 2005-10-21 | 2011-06-15 | Novartis AG | Combination of a renin-inhibitor and an anti-dyslipidemic agent and/or an antiobesity agent |
| EP2348016A1 (en) | 2006-06-09 | 2011-07-27 | Action Pharma A/S | Phenyl pyrrole aminoguanidine derivatives |
| WO2011095581A1 (en) | 2010-02-05 | 2011-08-11 | Intervet International B.V. | S piroindoline compounds for use as anthelminthi cs |
| WO2011106273A1 (en) | 2010-02-25 | 2011-09-01 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2011137024A1 (en) | 2010-04-26 | 2011-11-03 | Merck Sharp & Dohme Corp. | Novel spiropiperidine prolylcarboxypeptidase inhibitors |
| WO2011143057A1 (en) | 2010-05-11 | 2011-11-17 | Merck Sharp & Dohme Corp. | Novel prolylcarboxypeptidase inhibitors |
| WO2011156246A1 (en) | 2010-06-11 | 2011-12-15 | Merck Sharp & Dohme Corp. | Novel prolylcarboxypeptidase inhibitors |
| US8148429B2 (en) | 2000-08-07 | 2012-04-03 | Anamar Ab | Use of benzylideneaminoguanidines and hydroxyguanidines as melanocortin receptor ligands |
| US8247453B2 (en) | 2008-02-21 | 2012-08-21 | Janssen Pharmaceutica, Nv | Methods for the treatment of dermatological disorders |
| WO2012116145A1 (en) | 2011-02-25 | 2012-08-30 | Merck Sharp & Dohme Corp. | Novel cyclic azabenzimidazole derivatives useful as anti-diabetic agents |
| US8263605B2 (en) | 2006-02-22 | 2012-09-11 | Vertex Pharmaceutical Incorporated | Modulators of muscarinic receptors |
| US8299058B2 (en) | 2003-12-12 | 2012-10-30 | Syngenta Crop Protection Llc | Spiro-condensed indoline derivatives as pesticides |
| US8309567B2 (en) | 2003-12-12 | 2012-11-13 | Syngenta Crop Protection Llc | Spiroindoline derivatives having insecticidal properties |
| WO2013017678A1 (en) | 2011-08-04 | 2013-02-07 | Intervet International B.V. | Novel spiroindoline compounds |
| WO2013138352A1 (en) | 2012-03-15 | 2013-09-19 | Synergy Pharmaceuticals Inc. | Formulations of guanylate cyclase c agonists and methods of use |
| WO2014022528A1 (en) | 2012-08-02 | 2014-02-06 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US8648073B2 (en) | 2009-12-30 | 2014-02-11 | Fochon Pharma, Inc. | Certain dipeptidyl peptidase inhibitors |
| EP2698157A1 (en) | 2006-09-22 | 2014-02-19 | Merck Sharp & Dohme Corp. | Method of treatment using fatty acid synthesis inhibitors |
| WO2014130608A1 (en) | 2013-02-22 | 2014-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2014139388A1 (en) | 2013-03-14 | 2014-09-18 | Merck Sharp & Dohme Corp. | Novel indole derivatives useful as anti-diabetic agents |
| WO2014151206A1 (en) | 2013-03-15 | 2014-09-25 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase and their uses |
| WO2014151200A2 (en) | 2013-03-15 | 2014-09-25 | Synergy Pharmaceuticals Inc. | Compositions useful for the treatment of gastrointestinal disorders |
| EP2810951A2 (en) | 2008-06-04 | 2014-12-10 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase useful for the treatment of gastrointestinal disorders, inflammation, cancer and other disorders |
| WO2014197720A2 (en) | 2013-06-05 | 2014-12-11 | Synergy Pharmaceuticals, Inc. | Ultra-pure agonists of guanylate cyclase c, method of making and using same |
| WO2015051725A1 (en) | 2013-10-08 | 2015-04-16 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| WO2015054500A2 (en) | 2013-10-09 | 2015-04-16 | Synergy Pharmaceuticals, Inc. | Agonists of guanylate cyclase useful for downregulation of pro-inflammatory cytokines |
| US9018395B2 (en) | 2011-01-27 | 2015-04-28 | Université de Montréal | Pyrazolopyridine and pyrazolopyrimidine derivatives as melanocortin-4 receptor modulators |
| EP2881391A1 (en) | 2013-12-05 | 2015-06-10 | Bayer Pharma Aktiengesellschaft | Spiroindoline carbocycle derivatives and pharmaceutical compositions thereof |
| EP2933265A2 (en) | 2005-06-03 | 2015-10-21 | Amicus Therapeutics, Inc. | Pharmacological chaperones for treating obesity |
| WO2016030534A1 (en) | 2014-08-29 | 2016-03-03 | Tes Pharma S.R.L. | INHIBITORS OF α-AMINO-β-CARBOXYMUCONIC ACID SEMIALDEHYDE DECARBOXYLASE |
| EP2998314A1 (en) | 2007-06-04 | 2016-03-23 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase useful for the treatment of gastrointestinal disorders, inflammation, cancer and other disorders |
| US9468639B2 (en) | 2001-10-20 | 2016-10-18 | Sprout Pharmaceuticals, Inc. | Treating sexual desire disorders with flibanserin |
| US9546141B2 (en) | 2008-12-15 | 2017-01-17 | Sprout Pharmaceuticals, Inc. | Salts |
| US9688660B2 (en) | 2015-10-30 | 2017-06-27 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| US9730927B2 (en) | 2005-08-03 | 2017-08-15 | Sprout Pharmaceuticals, Inc. | Use of flibanserin in the treatment of obesity |
| US9763936B2 (en) | 2006-06-30 | 2017-09-19 | Sprout Pharmaceuticals, Inc. | Flibanserin for the treatment of urinary incontinence and related diseases |
| US9802935B2 (en) | 2015-10-30 | 2017-10-31 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| US9808457B2 (en) | 2015-10-30 | 2017-11-07 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| EP3241839A1 (en) | 2008-07-16 | 2017-11-08 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase useful for the treatment of gastrointestinal, inflammation, cancer and other disorders |
| WO2018069532A1 (en) | 2016-10-14 | 2018-04-19 | Tes Pharma S.R.L. | Inhibitors of alpha-amino-beta-carboxymuconic acid semialdehyde decarboxylase |
| WO2018106518A1 (en) | 2016-12-06 | 2018-06-14 | Merck Sharp & Dohme Corp. | Antidiabetic heterocyclic compounds |
| WO2018118670A1 (en) | 2016-12-20 | 2018-06-28 | Merck Sharp & Dohme Corp. | Antidiabetic spirochroman compounds |
| US10166230B2 (en) | 2007-09-12 | 2019-01-01 | Sprout Pharmaceuticals Inc. | Treatment of vasomotor symptoms |
| US10294214B2 (en) | 2016-06-07 | 2019-05-21 | Vanderbilt University | Positive allosteric modulators of human melanocortin-4 receptor |
| WO2020104456A1 (en) | 2018-11-20 | 2020-05-28 | Tes Pharma S.R.L | INHIBITORS OF α-AMINO-β-CARBOXYMUCONIC ACID SEMIALDEHYDE DECARBOXYLASE |
| US10675280B2 (en) | 2001-10-20 | 2020-06-09 | Sprout Pharmaceuticals, Inc. | Treating sexual desire disorders with flibanserin |
| WO2021133563A1 (en) * | 2019-12-23 | 2021-07-01 | Crinetics Pharmaceuticals, Inc. | Spirocyclic piperidine melanocortin subtype-2 receptor (mc2r) antagonists and uses thereof |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008056687A1 (en) * | 2006-11-09 | 2008-05-15 | Daiichi Sankyo Company, Limited | Novel spiropiperidine derivative |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996005203A1 (en) * | 1994-08-08 | 1996-02-22 | Merck Sharp & Dohme Limited | Spiro-substituted azacyclic derivatives and their use as therapeutic agents |
| US5578593A (en) * | 1992-12-11 | 1996-11-26 | Merck & Co., Inc. | Spiro piperidines and homologs promote release of growth hormone |
| US5731408A (en) * | 1995-04-10 | 1998-03-24 | Arizona Board Of Regents On Behalf Of The University Of Arizona | Peptides having potent antagonist and agonist bioactivities at melanocortin receptors |
-
1999
- 1999-06-10 CA CA002334551A patent/CA2334551A1/en not_active Abandoned
- 1999-06-10 EP EP99930220A patent/EP1085869A4/en not_active Withdrawn
- 1999-06-10 AU AU46801/99A patent/AU742425B2/en not_active Ceased
- 1999-06-10 JP JP2000553071A patent/JP2002517444A/en not_active Withdrawn
- 1999-06-10 WO PCT/US1999/013252 patent/WO1999064002A1/en not_active Application Discontinuation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5578593A (en) * | 1992-12-11 | 1996-11-26 | Merck & Co., Inc. | Spiro piperidines and homologs promote release of growth hormone |
| WO1996005203A1 (en) * | 1994-08-08 | 1996-02-22 | Merck Sharp & Dohme Limited | Spiro-substituted azacyclic derivatives and their use as therapeutic agents |
| US5731408A (en) * | 1995-04-10 | 1998-03-24 | Arizona Board Of Regents On Behalf Of The University Of Arizona | Peptides having potent antagonist and agonist bioactivities at melanocortin receptors |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1085869A4 * |
Cited By (231)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6350760B1 (en) | 1999-06-04 | 2002-02-26 | Merck & Co., Inc. | Substituted piperidines as melanocortin-4 receptor agonists |
| US7473760B2 (en) | 1999-06-29 | 2009-01-06 | Palatin Technologies, Inc. | Cyclic peptide compositions for treatment of sexual dysfunction |
| US7897721B2 (en) | 1999-06-29 | 2011-03-01 | Palatin Technologies, Inc. | Cyclic peptide compositions for treatment of sexual dysfunction |
| US6579968B1 (en) | 1999-06-29 | 2003-06-17 | Palatin Technologies, Inc. | Compositions and methods for treatment of sexual dysfunction |
| WO2001000224A1 (en) * | 1999-06-29 | 2001-01-04 | Palatin Technologies Inc. | Compositions and methods for treatment of sexual dysfunction |
| US7375125B2 (en) | 1999-08-04 | 2008-05-20 | Ore Pharmaceuticals, Inc. | Melanocortin-4 receptor binding compounds and methods of use thereof |
| WO2001010842A3 (en) * | 1999-08-04 | 2001-08-16 | Millennium Pharm Inc | Melanocortin-4 receptor binding compounds and methods of use thereof |
| US6699873B1 (en) | 1999-08-04 | 2004-03-02 | Millennium Pharmaceuticals, Inc. | Melanocortin-4 receptor binding compounds and methods of use thereof |
| AU2004202804B2 (en) * | 1999-08-04 | 2009-01-29 | Ore Pharmaceuticals Inc. | Melanocortin-4 Receptor Binding Compounds and Methods of use thereof |
| WO2001010842A2 (en) * | 1999-08-04 | 2001-02-15 | Millennium Pharmaceuticals, Inc. | Melanocortin-4 receptor binding compounds and methods of use thereof |
| WO2001055107A3 (en) * | 2000-01-28 | 2002-01-17 | Melacure Therapeutics Ab | Aromatic amines and amides acting on the melanocortin receptors |
| WO2001055109A1 (en) * | 2000-01-28 | 2001-08-02 | Melacure Therapeutics Ab | Aromatic amides acting on melanocortin receptors |
| WO2001055107A2 (en) * | 2000-01-28 | 2001-08-02 | Melacure Therapeutics Ab | Aromatic amines and amides acting on the melanocortin receptors |
| US6472398B1 (en) | 2000-03-23 | 2002-10-29 | Merck & Co., Inc. | Spiropiperidine derivatives as melanocortin receptor agonists |
| EP1268000A1 (en) * | 2000-03-23 | 2003-01-02 | Merck & Co., Inc. | Spiropiperidine derivatives as melanocortin receptor agonists |
| EP1268449A1 (en) * | 2000-03-23 | 2003-01-02 | Merck & Co., Inc. | Substituted piperidines as melanocortin receptor agonists |
| EP1268000A4 (en) * | 2000-03-23 | 2004-12-29 | Merck & Co Inc | Spiropiperidine derivatives as melanocortin receptor agonists |
| EP1268449A4 (en) * | 2000-03-23 | 2004-09-15 | Merck & Co Inc | Substituted piperidines as melanocortin receptor agonists |
| US6458790B2 (en) | 2000-03-23 | 2002-10-01 | Merck & Co., Inc. | Substituted piperidines as melanocortin receptor agonists |
| WO2001074844A2 (en) * | 2000-04-04 | 2001-10-11 | F. Hoffmann-La Roche Ag | Selective linear peptides with melanocortin-4 receptor (mc4-r) agonist activity |
| US6600015B2 (en) | 2000-04-04 | 2003-07-29 | Hoffmann-La Roche Inc. | Selective linear peptides with melanocortin-4 receptor (MC4-R) agonist activity |
| WO2001074844A3 (en) * | 2000-04-04 | 2002-06-13 | Hoffmann La Roche | Selective linear peptides with melanocortin-4 receptor (mc4-r) agonist activity |
| EP1289526A1 (en) * | 2000-05-30 | 2003-03-12 | Merck & Co., Inc. | Melanocortin receptor agonists |
| EP1289526A4 (en) * | 2000-05-30 | 2005-03-16 | Merck & Co Inc | Melanocortin receptor agonists |
| EP1295608A4 (en) * | 2000-06-27 | 2004-03-31 | Taisho Pharmaceutical Co Ltd | Remedial agent for anxiety neurosis or depression and piperazine derivative |
| EP1295608A1 (en) * | 2000-06-27 | 2003-03-26 | Taisho Pharmaceutical Co., Ltd | Remedial agent for anxiety neurosis or depression and piperazine derivative |
| JP5002881B2 (en) * | 2000-06-27 | 2012-08-15 | 大正製薬株式会社 | Anxiety or depression treatment and piperazine derivatives |
| US6949552B2 (en) | 2000-06-27 | 2005-09-27 | Taisho Pharmaceutical Co., Ltd. | Remedial agent for anxiety neurosis or depression and piperazine derivative |
| US7176279B2 (en) | 2000-06-28 | 2007-02-13 | Palatin Technologies, Inc. | Cyclic peptide compositions and methods for treatment of sexual dysfunction |
| WO2002000654A1 (en) * | 2000-06-28 | 2002-01-03 | Pfizer Products Inc. | Melanocortin receptor ligands |
| US8309609B2 (en) | 2000-08-07 | 2012-11-13 | Anamar Ab | Use of benzylideneaminoguanidines and hydroxyguanidines as melanocortin receptor ligands |
| US8148429B2 (en) | 2000-08-07 | 2012-04-03 | Anamar Ab | Use of benzylideneaminoguanidines and hydroxyguanidines as melanocortin receptor ligands |
| US8410174B2 (en) | 2000-08-07 | 2013-04-02 | Anamar Ab | Method for treating arthritis |
| US9227927B2 (en) | 2000-08-07 | 2016-01-05 | Anamar Ab | Method of treating inflammation |
| US7153881B2 (en) | 2000-08-07 | 2006-12-26 | Acure Pharma Ab | Compounds acting as melanocortin receptor ligands |
| KR100911888B1 (en) | 2000-08-07 | 2009-08-11 | 아큐어 파르마 아베 | Compounds acting as melanocortin receptor ligands |
| WO2002012178A1 (en) * | 2000-08-07 | 2002-02-14 | Melacure Therapeutics Ab | Compounds acting as melanocortin receptor ligands |
| US6767915B2 (en) | 2000-08-23 | 2004-07-27 | Merck & Co., Inc. | Substituted piperidines as melanocortin receptor agonists |
| US6995269B2 (en) | 2000-08-31 | 2006-02-07 | Chiron Corporation | Guanidinobenzamides |
| US6638927B2 (en) | 2000-08-31 | 2003-10-28 | Chiron Corporation | Guanidinobenzamides |
| WO2002018327A2 (en) * | 2000-08-31 | 2002-03-07 | Chiron Corporation | Guanidinobenzamides as mc4-r agonists |
| WO2002018327A3 (en) * | 2000-08-31 | 2002-08-08 | Chiron Corp | Guanidinobenzamides as mc4-r agonists |
| US7186715B2 (en) | 2001-01-08 | 2007-03-06 | Eli Lilly And Company | Piperazine- and piperidine-derivatives as melanocortin receptor agonists |
| WO2002059095A1 (en) * | 2001-01-23 | 2002-08-01 | Eli Lilly And Company | Melanocortin receptor agonists |
| US7291619B2 (en) | 2001-01-23 | 2007-11-06 | Eli Lilly And Company | Melanocortin receptor agonists |
| US7157463B2 (en) | 2001-01-23 | 2007-01-02 | Eli Lilly And Company | Substituted piperidines/piperazines as melanocortin receptor agonists |
| US7169777B2 (en) | 2001-01-23 | 2007-01-30 | Eli Lilly And Company | Melanocortin receptor agonists |
| WO2002059107A1 (en) * | 2001-01-23 | 2002-08-01 | Eli Lilly And Company | Substituted piperidines/piperazines as melanocortin receptor agonists |
| WO2002059108A1 (en) * | 2001-01-23 | 2002-08-01 | Eli Lilly And Company | Melanocortin receptor agonists |
| WO2002062766A2 (en) * | 2001-02-07 | 2002-08-15 | Millennium Pharmaceuticals, Inc. | Melanocortin-4 receptor binding compounds and methods of use thereof |
| WO2002062766A3 (en) * | 2001-02-07 | 2002-10-03 | Millennium Pharm Inc | Melanocortin-4 receptor binding compounds and methods of use thereof |
| US7307063B2 (en) | 2001-02-13 | 2007-12-11 | Palatin Technologies, Inc. | Melanocortin metallopeptides for treatment of sexual dysfunction |
| US6818658B2 (en) | 2001-02-28 | 2004-11-16 | Merck & Co., Inc. | Acylated piperidine derivatives as melanocortin-4 receptor agonists |
| US7700778B2 (en) | 2001-02-28 | 2010-04-20 | Merck Sharp & Dohme Corp. | Acylated piperidine derivatives as melanocortin-4 receptor agonists |
| US7012084B2 (en) | 2001-02-28 | 2006-03-14 | Merck & Co., Inc. | Acylated piperidine derivatives as melanocortin-4 receptor agonists |
| EP1363898A4 (en) * | 2001-03-02 | 2004-12-08 | Bristol Myers Squibb Co | Compounds useful as modulators of melanocortin receptors and pharmaceutical compositions comprising same |
| EP1363898A1 (en) * | 2001-03-02 | 2003-11-26 | Bristol-Myers Squibb Company | Compounds useful as modulators of melanocortin receptors and pharmaceutical compositions comprising same |
| US6713487B2 (en) | 2001-03-02 | 2004-03-30 | Bristol-Myers Squibb Co. | Compounds useful as modulators of melanocortin receptors and pharmaceutical compositions comprising same |
| US7067525B2 (en) | 2001-03-02 | 2006-06-27 | Bristol-Myers Squibb Co. | Compounds useful as modulators of Melanocortin Receptors and pharmaceutical compositions comprising same |
| US6979691B2 (en) | 2001-03-02 | 2005-12-27 | Bristol-Myers Squibb Company | Compounds useful as modulators of melanocortin receptors and pharmaceutical compositions comprising same |
| US7456183B2 (en) | 2001-04-09 | 2008-11-25 | Novartis Vaccines And Diagnostics, Inc. | Guanidino compounds |
| US6960582B2 (en) | 2001-04-09 | 2005-11-01 | Chiron Corporation | Guanidino compounds |
| US7189727B2 (en) | 2001-04-09 | 2007-03-13 | Chiron Corporation | Guanidino compounds |
| US6716840B2 (en) | 2001-04-09 | 2004-04-06 | Chiron Corporation | Guanidino compounds |
| US7087759B2 (en) | 2001-04-25 | 2006-08-08 | The Procter & Gamble Company | Melanocortin receptor ligands |
| US6911447B2 (en) | 2001-04-25 | 2005-06-28 | The Procter & Gamble Company | Melanocortin receptor ligands |
| US7125885B2 (en) | 2001-05-04 | 2006-10-24 | Amgen Inc. | Fused heterocyclic compounds |
| WO2002094825A1 (en) * | 2001-05-22 | 2002-11-28 | Banyu Pharmaceutical Co., Ltd. | Novel spiropiperidine derivative |
| US7115628B2 (en) | 2001-07-18 | 2006-10-03 | Merck & Co., Inc. | Bridged piperidine derivatives as melanocortin receptor agonists |
| US7115607B2 (en) | 2001-07-25 | 2006-10-03 | Amgen Inc. | Substituted piperazinyl amides and methods of use |
| US7560460B2 (en) | 2001-07-25 | 2009-07-14 | Amgen Inc. | Substituted piperazines and methods of use |
| WO2003009850A1 (en) * | 2001-07-25 | 2003-02-06 | Amgen Inc. | Substituted piperazines as modulators of the melanocortin receptor |
| EP1425029A1 (en) * | 2001-08-10 | 2004-06-09 | Palatin Technologies, Inc. | Peptidomimetics of biologically active metallopeptides |
| EP1425029A4 (en) * | 2001-08-10 | 2006-06-07 | Palatin Technologies Inc | Peptidomimetics of biologically active metallopeptides |
| US7326707B2 (en) | 2001-08-10 | 2008-02-05 | Palatin Technologies Incorporated | Bicyclic melanocortin-specific compounds |
| EP1285658A3 (en) * | 2001-08-21 | 2004-01-28 | Pfizer Productors Inc. | Treatments for female sexual dysfunction |
| EP1285658A2 (en) * | 2001-08-21 | 2003-02-26 | Pfizer Productors Inc. | Treatments for female sexual dysfunction |
| KR20030027439A (en) * | 2001-09-28 | 2003-04-07 | 주식회사 엘지생명과학 | Melanocortin receptor agonists |
| US6916812B2 (en) | 2001-10-09 | 2005-07-12 | Bristol-Myers Squibb Company | Alpha-aminoamide derivatives as melanocortin agonists |
| US9782403B2 (en) | 2001-10-20 | 2017-10-10 | Sprout Pharmaceuticals, Inc. | Treating sexual desire disorders with flibanserin |
| US9468639B2 (en) | 2001-10-20 | 2016-10-18 | Sprout Pharmaceuticals, Inc. | Treating sexual desire disorders with flibanserin |
| US10675280B2 (en) | 2001-10-20 | 2020-06-09 | Sprout Pharmaceuticals, Inc. | Treating sexual desire disorders with flibanserin |
| US11058683B2 (en) | 2001-10-20 | 2021-07-13 | Sprout Pharmaceuticals, Inc. | Treating sexual desire disorders with flibanserin |
| KR20030035589A (en) * | 2001-10-31 | 2003-05-09 | 주식회사 엘지생명과학 | Melanocortin receptor agonists |
| KR20030035592A (en) * | 2001-10-31 | 2003-05-09 | 주식회사 엘지생명과학 | Melanocortin receptor agonists |
| US7049331B2 (en) | 2001-11-08 | 2006-05-23 | Ortho-Mcneil Pharmaceutical, Inc. | 1,2,4-thiadiazole derivatives as melanocortin receptor modulators |
| US8501791B2 (en) | 2001-11-08 | 2013-08-06 | Ortho-Mcneil Pharamaceutical, Inc. | 1,2,4-thiadiazolium derivatives as melanocortin receptor modulators |
| US7786311B2 (en) | 2001-11-08 | 2010-08-31 | Ortho-Mcneil Pharmaceutical, Inc. | 1,2,4-thiadiazolium derivatives as melanocortin receptor modulators |
| US7319107B2 (en) | 2001-11-08 | 2008-01-15 | Johnson & Johnson Consumer Companies, Inc. | 1,2,4-thiadiazolium derivatives as melanocortin receptor modulators |
| US7375123B2 (en) | 2001-11-08 | 2008-05-20 | Ortho-Mcneil Pharmaceutical, Inc. | 1,2,4-thiadiazole derivatives as melanocortin receptor modulators |
| US7314879B2 (en) | 2002-01-23 | 2008-01-01 | Eli Lilly And Company | Melanocortin receptor agonists |
| WO2003061660A1 (en) * | 2002-01-23 | 2003-07-31 | Eli Lilly And Company | Melanocortin receptor agonists |
| WO2003066587A3 (en) * | 2002-02-04 | 2004-03-11 | Chiron Corp | Piperidine or pyrrolidine derivatives having an aminoacyl substituted side chain as melancortin-4 receptor agonists |
| WO2003066587A2 (en) * | 2002-02-04 | 2003-08-14 | Chiron Corporation | Piperidine or pyrrolidine derivatives having an aminoacyl substituted side chain as melancortin-4 receptor agonists |
| US7026335B2 (en) | 2002-04-30 | 2006-04-11 | The Procter & Gamble Co. | Melanocortin receptor ligands |
| WO2003094918A1 (en) * | 2002-05-10 | 2003-11-20 | Neurocrine Biosciences, Inc. | Substituted piperazine as melanocortin receptors ligands |
| US7858641B2 (en) | 2002-05-23 | 2010-12-28 | Novartis Vaccines And Diagnostics, Inc. | Substituted dihydroisoquinolinone compounds |
| US7858631B2 (en) | 2002-05-23 | 2010-12-28 | Novartis Vaccines And Diagnostics, Inc. | Substituted pyrido [2,3-d] pyrimidinone compounds |
| US7034033B2 (en) | 2002-05-23 | 2006-04-25 | Chiron Corporation | Substituted quinazolinone compounds |
| EP1880996A1 (en) | 2002-06-14 | 2008-01-23 | Syngenta Limited | Spiroindolinepiperidine derivatives |
| WO2004002986A2 (en) | 2002-06-28 | 2004-01-08 | Banyu Pharmaceutical Co., Ltd. | Novel benzimidazole derivatives |
| US7795378B2 (en) | 2002-07-09 | 2010-09-14 | Palatin Technologies, Inc. | Peptide compositions for treatment of sexual dysfunction |
| US7414057B2 (en) | 2002-09-11 | 2008-08-19 | Merck & Co., Inc. | Piperazine urea derivatives as melanocortin-4 receptor agonists |
| US7045527B2 (en) | 2002-09-24 | 2006-05-16 | Amgen Inc. | Piperidine derivatives |
| US7132539B2 (en) | 2002-10-23 | 2006-11-07 | The Procter & Gamble Company | Melanocortin receptor ligands |
| US7253179B2 (en) | 2002-11-06 | 2007-08-07 | Amgen Inc. | Fused heterocyclic compounds |
| US7576081B2 (en) | 2002-12-10 | 2009-08-18 | Pfizer Inc. | Morpholine dopamine agonists |
| US7323462B2 (en) | 2002-12-10 | 2008-01-29 | Pfizer Inc. | Morpholine dopamine agonists |
| US7902188B2 (en) | 2002-12-10 | 2011-03-08 | Pfizer Inc. | Morpholine dopamine agonists |
| US7160886B2 (en) | 2003-03-03 | 2007-01-09 | Merck & Co., Inc. | Acylated piperazine derivatives as melanocortin-4 receptor agonists |
| WO2004083209A1 (en) * | 2003-03-20 | 2004-09-30 | Santhera Pharmaceuticals (Schweiz) Gmbh | Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators |
| US7276520B2 (en) | 2003-03-26 | 2007-10-02 | Merck & Co., Inc. | Bicyclic piperidine derivatives as melanocortin-4 receptor agonists |
| US7329673B2 (en) | 2003-04-04 | 2008-02-12 | Merck & Co., Inc. | Acylated spiropiperidine derivatives as melanocortin-4 receptor agonists |
| US7049323B2 (en) | 2003-04-25 | 2006-05-23 | Bristol-Myers Squibb Company | Amidoheterocycles as modulators of the melanocortin-4 receptor |
| US7625909B2 (en) | 2003-05-23 | 2009-12-01 | Novartis Vaccines And Diagnostics, Inc. | Substituted quinazolinone compounds |
| WO2005028438A1 (en) | 2003-09-22 | 2005-03-31 | Banyu Pharmaceutical Co., Ltd. | Novel piperidine derivative |
| US7368453B2 (en) | 2003-11-19 | 2008-05-06 | Chiron Corporation | Quinazolinone compounds with reduced bioaccumulation |
| US7420059B2 (en) | 2003-11-20 | 2008-09-02 | Bristol-Myers Squibb Company | HMG-CoA reductase inhibitors and method |
| US8299058B2 (en) | 2003-12-12 | 2012-10-30 | Syngenta Crop Protection Llc | Spiro-condensed indoline derivatives as pesticides |
| WO2005058836A1 (en) * | 2003-12-12 | 2005-06-30 | Syngenta Participations Ag | Insecticidal spiroindane derivatives |
| US8354423B2 (en) | 2003-12-12 | 2013-01-15 | Syngenta Crop Protection Llc | Insecticidal spironindane derivatives |
| US8309567B2 (en) | 2003-12-12 | 2012-11-13 | Syngenta Crop Protection Llc | Spiroindoline derivatives having insecticidal properties |
| US8846907B2 (en) | 2003-12-12 | 2014-09-30 | Syngenta Crop Protection Llc | Spiro-condensed indoline derivatives as pesticides |
| EP2088154A1 (en) | 2004-03-09 | 2009-08-12 | Ironwood Pharmaceuticals, Inc. | Methods and compositions for the treatment of gastrointestinal disorders |
| WO2005097759A1 (en) | 2004-03-29 | 2005-10-20 | Merck & Co., Inc. | Diaryltriazoles as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| EP2305352A1 (en) | 2004-04-02 | 2011-04-06 | Merck Sharp & Dohme Corp. | 5-alpha-reductase inhibitors for use in the treatment of men with metabolic and anthropometric disorders |
| US7618987B2 (en) | 2004-07-19 | 2009-11-17 | Merck & Co., Inc. | Acylated piperidine derivatives as melanocortin 4-receptor agonists |
| WO2006017542A1 (en) | 2004-08-06 | 2006-02-16 | Merck & Co., Inc. | Sulfonyl compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2006129826A1 (en) | 2005-05-30 | 2006-12-07 | Banyu Pharmaceutical Co., Ltd. | Novel piperidine derivative |
| EP2933265A2 (en) | 2005-06-03 | 2015-10-21 | Amicus Therapeutics, Inc. | Pharmacological chaperones for treating obesity |
| US10335407B2 (en) | 2005-08-03 | 2019-07-02 | Sprout Pharmaceuticals, Inc. | Use of flibanserin in the treatment of obesity |
| US9730927B2 (en) | 2005-08-03 | 2017-08-15 | Sprout Pharmaceuticals, Inc. | Use of flibanserin in the treatment of obesity |
| US10874668B2 (en) | 2005-08-03 | 2020-12-29 | Sprout Pharmaceuticals, Inc. | Use of Flibanserin in the treatment of obesity |
| WO2007018248A1 (en) | 2005-08-10 | 2007-02-15 | Banyu Pharmaceutical Co., Ltd. | Pyridone compound |
| WO2007024004A1 (en) | 2005-08-24 | 2007-03-01 | Banyu Pharmaceutical Co., Ltd. | Phenylpyridone derivative |
| WO2007029847A1 (en) | 2005-09-07 | 2007-03-15 | Banyu Pharmaceutical Co., Ltd. | Bicyclic aromatic substituted pyridone derivative |
| WO2007041052A2 (en) | 2005-09-29 | 2007-04-12 | Merck & Co., Inc. | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| AU2006297443B2 (en) * | 2005-09-29 | 2010-08-12 | Merck Sharp & Dohme Corp. | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| JP2009510067A (en) * | 2005-09-29 | 2009-03-12 | メルク エンド カムパニー インコーポレーテッド | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| US8293900B2 (en) | 2005-09-29 | 2012-10-23 | Merck Sharp & Dohme Corp | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| WO2007041052A3 (en) * | 2005-09-29 | 2007-05-31 | Merck & Co Inc | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| US7652024B2 (en) | 2005-10-18 | 2010-01-26 | Merck Sharp & Dohme Corp. | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| EP2332526A2 (en) | 2005-10-21 | 2011-06-15 | Novartis AG | Combination of a renin-inhibitor and an anti-dyslipidemic agent and/or an antiobesity agent |
| WO2007049798A1 (en) | 2005-10-27 | 2007-05-03 | Banyu Pharmaceutical Co., Ltd. | Novel benzoxathiin derivative |
| WO2007055418A1 (en) | 2005-11-10 | 2007-05-18 | Banyu Pharmaceutical Co., Ltd. | Aza-substituted spiro derivative |
| EP1801098A1 (en) | 2005-12-16 | 2007-06-27 | Merck Sante | 2-Adamantylurea derivatives as selective 11B-HSD1 inhibitors |
| US7858635B2 (en) | 2005-12-22 | 2010-12-28 | Vertex Pharmaceuticals Incorporated | Spiro compounds as modulators of muscarinic receptors |
| WO2007100664A3 (en) * | 2006-02-22 | 2008-03-06 | Vertex Pharma | Modulators of muscarinic receptors |
| US8263605B2 (en) | 2006-02-22 | 2012-09-11 | Vertex Pharmaceutical Incorporated | Modulators of muscarinic receptors |
| US8058306B2 (en) | 2006-06-09 | 2011-11-15 | Action Pharma A/S | Phenyl pyrrole aminoguanidine derivatives |
| EP2348016A1 (en) | 2006-06-09 | 2011-07-27 | Action Pharma A/S | Phenyl pyrrole aminoguanidine derivatives |
| US7858790B2 (en) | 2006-06-29 | 2010-12-28 | Vertex Pharmaceuticals Incorporated | Modulators of muscarinic receptors |
| US10004731B2 (en) | 2006-06-30 | 2018-06-26 | Sprout Pharmaceuticals, Inc. | Flibanserin for the treatment of urinary incontinence and related diseases |
| US9763936B2 (en) | 2006-06-30 | 2017-09-19 | Sprout Pharmaceuticals, Inc. | Flibanserin for the treatment of urinary incontinence and related diseases |
| US7696201B2 (en) | 2006-08-15 | 2010-04-13 | Vertex Pharmaceuticals Incorporated | Modulators of muscarinic receptors |
| EP2946778A1 (en) | 2006-09-22 | 2015-11-25 | Merck Sharp & Dohme Corp. | Method of treatment using fatty acid synthesis inhibitors |
| EP2698157A1 (en) | 2006-09-22 | 2014-02-19 | Merck Sharp & Dohme Corp. | Method of treatment using fatty acid synthesis inhibitors |
| WO2008039418A2 (en) * | 2006-09-27 | 2008-04-03 | Merck & Co., Inc. | Acylated piperidine derivatives as melanocortin-4 receptor modulators |
| WO2008039863A2 (en) * | 2006-09-27 | 2008-04-03 | Braincells, Inc. | Composition comprising a melanocortin receptor (mcr) modulating agent alone or in combination with a second neurogenic agent for treating nervous system disorders |
| WO2008039418A3 (en) * | 2006-09-27 | 2008-10-02 | Merck & Co Inc | Acylated piperidine derivatives as melanocortin-4 receptor modulators |
| WO2008039863A3 (en) * | 2006-09-27 | 2009-01-08 | Braincells Inc | Composition comprising a melanocortin receptor (mcr) modulating agent alone or in combination with a second neurogenic agent for treating nervous system disorders |
| WO2008038692A1 (en) | 2006-09-28 | 2008-04-03 | Banyu Pharmaceutical Co., Ltd. | Diaryl ketimine derivative |
| WO2008071980A1 (en) | 2006-12-14 | 2008-06-19 | Acure Pharma Ab | Novel aminoguanidines as melanocortin receptor ligands. |
| US8372878B2 (en) | 2006-12-14 | 2013-02-12 | Anamar Ab | Aminoguanidines as melanocortin receptor ligands |
| WO2008074384A1 (en) | 2006-12-21 | 2008-06-26 | Merck Patent Gmbh | 2-ADAMANTYL-BUTYRAMIDE DERIVATIVES AS SELECTIVE 11βETA-HSD1 INHIBITORS |
| WO2008120653A1 (en) | 2007-04-02 | 2008-10-09 | Banyu Pharmaceutical Co., Ltd. | Indoledione derivative |
| EP2998314A1 (en) | 2007-06-04 | 2016-03-23 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase useful for the treatment of gastrointestinal disorders, inflammation, cancer and other disorders |
| US10166230B2 (en) | 2007-09-12 | 2019-01-01 | Sprout Pharmaceuticals Inc. | Treatment of vasomotor symptoms |
| US8536226B2 (en) | 2008-02-21 | 2013-09-17 | Janssen Pharmaceutica, Nv | Methods for the treatment of dermatological disorders |
| US8247453B2 (en) | 2008-02-21 | 2012-08-21 | Janssen Pharmaceutica, Nv | Methods for the treatment of dermatological disorders |
| WO2009110510A1 (en) | 2008-03-06 | 2009-09-11 | 萬有製薬株式会社 | Alkylaminopyridine derivative |
| WO2009119726A1 (en) | 2008-03-28 | 2009-10-01 | 萬有製薬株式会社 | Diarylmethylamide derivative having antagonistic activity on melanin-concentrating hormone receptor |
| WO2009127321A1 (en) | 2008-04-18 | 2009-10-22 | Merck Patent Gmbh, | Benzofurane, benzothiophene, benzothiazol derivatives as fxr modulators |
| EP2110374A1 (en) | 2008-04-18 | 2009-10-21 | Merck Sante | Benzofurane, benzothiophene, benzothiazol derivatives as FXR modulators |
| EP2810951A2 (en) | 2008-06-04 | 2014-12-10 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase useful for the treatment of gastrointestinal disorders, inflammation, cancer and other disorders |
| WO2009154132A1 (en) | 2008-06-19 | 2009-12-23 | 萬有製薬株式会社 | Spirodiamine-diarylketoxime derivative |
| EP3241839A1 (en) | 2008-07-16 | 2017-11-08 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase useful for the treatment of gastrointestinal, inflammation, cancer and other disorders |
| WO2010013595A1 (en) | 2008-07-30 | 2010-02-04 | 萬有製薬株式会社 | (5-membered)-(5-membered) or (5-membered)-(6-membered) fused ring cycloalkylamine derivative |
| WO2010047982A1 (en) | 2008-10-22 | 2010-04-29 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2010051206A1 (en) | 2008-10-31 | 2010-05-06 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2010056717A1 (en) | 2008-11-17 | 2010-05-20 | Merck Sharp & Dohme Corp. | Substituted bicyclic amines for the treatment of diabetes |
| US9546141B2 (en) | 2008-12-15 | 2017-01-17 | Sprout Pharmaceuticals, Inc. | Salts |
| WO2011011508A1 (en) | 2009-07-23 | 2011-01-27 | Schering Corporation | Benzo-fused oxazepine compounds as stearoyl-coenzyme a delta-9 desaturase inhibitors |
| WO2011011506A1 (en) | 2009-07-23 | 2011-01-27 | Schering Corporation | Spirocyclic oxazepine compounds as stearoyl-coenzyme a delta-9 desaturase inhibitors |
| US8765728B2 (en) | 2009-11-16 | 2014-07-01 | Mellitech | [1,5]-diazocin derivatives |
| WO2011058193A1 (en) | 2009-11-16 | 2011-05-19 | Mellitech | [1,5]-diazocin derivatives |
| EP2923706A1 (en) | 2009-12-03 | 2015-09-30 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase useful for the treatment of hypercholesterolemia |
| WO2011069038A2 (en) | 2009-12-03 | 2011-06-09 | Synergy Pharmaceuticals, Inc. | Agonists of guanylate cyclase useful for the treatment of hypercholesterolemia, atherosclerosis, coronary heart disease, gallstone, obesity and other cardiovascular diseases |
| US8648073B2 (en) | 2009-12-30 | 2014-02-11 | Fochon Pharma, Inc. | Certain dipeptidyl peptidase inhibitors |
| US9340523B2 (en) | 2009-12-30 | 2016-05-17 | Fochon Pharma, Inc. | Certain dipeptidyl peptidase inhibitors |
| US9598412B2 (en) | 2010-02-05 | 2017-03-21 | Intervet Inc. | Spiroindoline compounds for use as anthelminthics |
| WO2011095581A1 (en) | 2010-02-05 | 2011-08-11 | Intervet International B.V. | S piroindoline compounds for use as anthelminthi cs |
| WO2011106273A1 (en) | 2010-02-25 | 2011-09-01 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2011137024A1 (en) | 2010-04-26 | 2011-11-03 | Merck Sharp & Dohme Corp. | Novel spiropiperidine prolylcarboxypeptidase inhibitors |
| WO2011143057A1 (en) | 2010-05-11 | 2011-11-17 | Merck Sharp & Dohme Corp. | Novel prolylcarboxypeptidase inhibitors |
| WO2011156246A1 (en) | 2010-06-11 | 2011-12-15 | Merck Sharp & Dohme Corp. | Novel prolylcarboxypeptidase inhibitors |
| US9018395B2 (en) | 2011-01-27 | 2015-04-28 | Université de Montréal | Pyrazolopyridine and pyrazolopyrimidine derivatives as melanocortin-4 receptor modulators |
| US9493456B2 (en) | 2011-01-27 | 2016-11-15 | Universite De Montreal | Pyrazolopyridine and pyrazolopyrimidine derivatives as melanocortin-4 receptor modulators |
| EP3243385A1 (en) | 2011-02-25 | 2017-11-15 | Merck Sharp & Dohme Corp. | Novel cyclic azabenzimidazole derivatives useful as anti-diabetic agents |
| WO2012116145A1 (en) | 2011-02-25 | 2012-08-30 | Merck Sharp & Dohme Corp. | Novel cyclic azabenzimidazole derivatives useful as anti-diabetic agents |
| WO2013017678A1 (en) | 2011-08-04 | 2013-02-07 | Intervet International B.V. | Novel spiroindoline compounds |
| WO2013138352A1 (en) | 2012-03-15 | 2013-09-19 | Synergy Pharmaceuticals Inc. | Formulations of guanylate cyclase c agonists and methods of use |
| EP4309673A2 (en) | 2012-03-15 | 2024-01-24 | Bausch Health Ireland Limited | Formulations of guanylate cyclase c agonists and methods of use |
| EP3708179A1 (en) | 2012-03-15 | 2020-09-16 | Bausch Health Ireland Limited | Formulations of guanylate cyclase c agonists and methods of use |
| WO2014022528A1 (en) | 2012-08-02 | 2014-02-06 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| WO2014130608A1 (en) | 2013-02-22 | 2014-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2014139388A1 (en) | 2013-03-14 | 2014-09-18 | Merck Sharp & Dohme Corp. | Novel indole derivatives useful as anti-diabetic agents |
| WO2014151200A2 (en) | 2013-03-15 | 2014-09-25 | Synergy Pharmaceuticals Inc. | Compositions useful for the treatment of gastrointestinal disorders |
| WO2014151206A1 (en) | 2013-03-15 | 2014-09-25 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase and their uses |
| WO2014197720A2 (en) | 2013-06-05 | 2014-12-11 | Synergy Pharmaceuticals, Inc. | Ultra-pure agonists of guanylate cyclase c, method of making and using same |
| WO2015051725A1 (en) | 2013-10-08 | 2015-04-16 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| WO2015054500A2 (en) | 2013-10-09 | 2015-04-16 | Synergy Pharmaceuticals, Inc. | Agonists of guanylate cyclase useful for downregulation of pro-inflammatory cytokines |
| EP2881391A1 (en) | 2013-12-05 | 2015-06-10 | Bayer Pharma Aktiengesellschaft | Spiroindoline carbocycle derivatives and pharmaceutical compositions thereof |
| US11254644B2 (en) | 2014-08-29 | 2022-02-22 | Tes Pharma S.R.L. | Inhibitors of alpha-amino-beta-carboxymuconic acid semialdehyde decarboxylase |
| US9708272B2 (en) | 2014-08-29 | 2017-07-18 | Tes Pharma S.R.L. | Inhibitors of α-amino-β-carboxymuconic acid semialdehyde decarboxylase |
| WO2016030534A1 (en) | 2014-08-29 | 2016-03-03 | Tes Pharma S.R.L. | INHIBITORS OF α-AMINO-β-CARBOXYMUCONIC ACID SEMIALDEHYDE DECARBOXYLASE |
| US10513499B2 (en) | 2014-08-29 | 2019-12-24 | Tes Pharma S.R.L. | Inhibitors of alpha-amino-beta-carboxymuconic acid semialdehyde decarboxylase |
| US9808457B2 (en) | 2015-10-30 | 2017-11-07 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| US9925178B2 (en) | 2015-10-30 | 2018-03-27 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| US10300056B2 (en) | 2015-10-30 | 2019-05-28 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| US10259812B2 (en) | 2015-10-30 | 2019-04-16 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| US10166226B2 (en) | 2015-10-30 | 2019-01-01 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| US9688660B2 (en) | 2015-10-30 | 2017-06-27 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| US9802935B2 (en) | 2015-10-30 | 2017-10-31 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| US10888561B2 (en) | 2015-10-30 | 2021-01-12 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| US10294214B2 (en) | 2016-06-07 | 2019-05-21 | Vanderbilt University | Positive allosteric modulators of human melanocortin-4 receptor |
| WO2018069532A1 (en) | 2016-10-14 | 2018-04-19 | Tes Pharma S.R.L. | Inhibitors of alpha-amino-beta-carboxymuconic acid semialdehyde decarboxylase |
| WO2018106518A1 (en) | 2016-12-06 | 2018-06-14 | Merck Sharp & Dohme Corp. | Antidiabetic heterocyclic compounds |
| WO2018118670A1 (en) | 2016-12-20 | 2018-06-28 | Merck Sharp & Dohme Corp. | Antidiabetic spirochroman compounds |
| WO2020104456A1 (en) | 2018-11-20 | 2020-05-28 | Tes Pharma S.R.L | INHIBITORS OF α-AMINO-β-CARBOXYMUCONIC ACID SEMIALDEHYDE DECARBOXYLASE |
| WO2021133563A1 (en) * | 2019-12-23 | 2021-07-01 | Crinetics Pharmaceuticals, Inc. | Spirocyclic piperidine melanocortin subtype-2 receptor (mc2r) antagonists and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2002517444A (en) | 2002-06-18 |
| AU4680199A (en) | 1999-12-30 |
| CA2334551A1 (en) | 1999-12-16 |
| EP1085869A1 (en) | 2001-03-28 |
| EP1085869A4 (en) | 2001-10-04 |
| AU742425B2 (en) | 2002-01-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6294534B1 (en) | Spiropiperidine derivatives as melanocortin receptor agonists | |
| AU742425B2 (en) | Spiropiperidine derivatives as melanocortin receptor agonists | |
| JP4336196B2 (en) | Crosslinked piperidine derivatives as melanocortin receptor agonists | |
| US6350760B1 (en) | Substituted piperidines as melanocortin-4 receptor agonists | |
| US6376509B2 (en) | Melanocortin receptor agonists | |
| US6458790B2 (en) | Substituted piperidines as melanocortin receptor agonists | |
| AU2001249281B2 (en) | Spiropiperidine derivatives as melanocortin receptor agonists | |
| US6767915B2 (en) | Substituted piperidines as melanocortin receptor agonists | |
| AU2001264977A1 (en) | Melanocortin receptor agonists | |
| AU2001288285A1 (en) | Substituted piperidines as melanocortin receptor agonists | |
| AU2002320494A1 (en) | Bridged piperidine derivatives as melanocortin receptor agonists | |
| AU2001249296A1 (en) | Substituted piperidines as melanocortin receptor agonists | |
| AU2001249281A1 (en) | Spiropiperidine derivatives as melanocortin receptor agonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AL AM AU AZ BA BB BG BR BY CA CN CU CZ EE GD GE HR HU ID IL IN IS JP KG KR KZ LC LK LR LT LV MD MG MK MN MX NO NZ PL RO RU SG SI SK TJ TM TR TT UA US UZ VN YU ZA |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 46801/99 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2334551 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2000 553071 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999930220 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999930220 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 46801/99 Country of ref document: AU |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1999930220 Country of ref document: EP |