WO1997049704A1 - N-[4-(heteroarylmethyl)phenyl]-heteroarylamines - Google Patents
N-[4-(heteroarylmethyl)phenyl]-heteroarylamines Download PDFInfo
- Publication number
- WO1997049704A1 WO1997049704A1 PCT/EP1997/003248 EP9703248W WO9749704A1 WO 1997049704 A1 WO1997049704 A1 WO 1997049704A1 EP 9703248 W EP9703248 W EP 9703248W WO 9749704 A1 WO9749704 A1 WO 9749704A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- alkyl
- amino
- aryl
- phenyl
- Prior art date
Links
- 0 CC1=C(C)N=C(C)*1 Chemical compound CC1=C(C)N=C(C)*1 0.000 description 6
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the present invention concerns N-[4-(heteroaryl-methyl)phenyl]-heteroarylamines, their N-oxides and addition salts; it further relates to processes for their preparation, and compositions comprising them.
- the compounds of the present invention are potent inhibitors of the retinoic acid-metabolism, and hence, their use as a medicine is also described.
- EP-A-0,260,744 published on March 23, 1988, discloses (IH-imidazol-l -ylmethyl) substituted benzimidazoles as inhibitors of the androgen formation from C2 1 -steroids, as inhibitors of the biosynthesis of thromboxane A2, and also having the capability to increase the excretion of ureic acid.
- EP-A-0,371 ,559 published on June 6, 1990, discloses said benzimidazoles and analogous benzotriazoles as potent suppressers of the plasma elimination of endogenously or exogenously administered retinoic acid.
- Retinoic acid is a key molecule in the regulation of growth and differentiation of epithelial tissues.
- RA is very rapidly metabolized by a series of enzymatic reactions, which results in its deactivation. Inhibition of RA-metabolism leads to enhanced RA levels in plasma and tissue. Therefore, compounds with such an inhibitory action, also called retinoic mimetic activity, have therapeutic and/or preventive potential in the field of dermatology and oncology.
- novel compounds of the present invention have retinoic mimetic activity and, moreover, show little or no endocrinological side-effects.
- the present invention is concerned with compounds of formula
- Ri represents hydrogen, hydroxy, C- ⁇ alkyl or aryl
- R2 represents hydrogen; Ci- ⁇ al yl; C3_ cycloalkyl; C2-8alkenyl; aryl; pyrrolidinyl optionally substituted with C ] -4 alkyl or C
- R3 represents hydrogen, C*-6alkyl, aryl or C
- Het represents an unsaturated heterocycle selected from lmidazolyl, t ⁇ azolyl, tetrazolyl and pyridinyl, each of said unsaturated heterocycles may optionally be substituted with ammo, mercapto, Ci ⁇ alkyl, C ⁇ 6 alkylth ⁇ o or aryl, represents an unsaturated mono- or bicychc heterocycle selected from the group consisting of pyridinyl, py ⁇ dazinyl, py ⁇ midinyl, pyrazmyl, qumolinyl, isoquinolmyl, pu ⁇ nyl, phtalazinyl, cinnoliny
- R 8 is hydrogen, C- ⁇ alkyl, aryl or arylCi ⁇ alkyl
- R4 and R ⁇ each independently represent hydrogen, hydroxy, halo, cyano, nitro, amino, formyl, carboxyl, mono- or d ⁇ (C
- each R 9 independently represents hydrogen, hydroxy, halo, nitro, amino, formyl, carboxyl, mono- or d C- ⁇ alkyDamino, C- ⁇ alkyloxy- carbonyl or aryl; and aryl represents phenyl or phenyl substituted with one, two or three substituents selected from hydroxy, halo, cyano, amino, mono- or Ci ⁇ alkyl, haloCi ⁇ alkyl, hydroxyCi- ⁇ alkyl, C ⁇ _6alkyloxy, formyl, carboxyl and
- Cj. ⁇ alkylcarbonyl; or two adjacent carbon atoms on said phenyl may be substituted by a single bivalent radical having the formula C-. ⁇ alkanediyl or
- halo is generic to fluoro, chloro, bro o and iodo
- C3_7cycloalkyl is generic to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl
- C2-8alkenyl defines straight and branch chained hydro ⁇ carbon radicals containing one double bond and having from 2 to 8 carbon atoms such as, for example, ethenyl, 1-propenyl, 2-butenyl, 2-pentenyl, 3-pentenyl, 3-methyl-2- butenyl, 3-hexenyl, 3-heptenyl, 2-octenyl and the like
- Ci ⁇ alkyl defines straight and branched chain saturated hydrocarbon radicals having from 1 to 4 carbon atoms such as, for example, methyl, ethyl, propyl, butyl, 1 -methylethyl, 2-methylpropyl, 2,
- Ci. ⁇ alkanediyl defines bivalent straight and branched chain saturated hydrocarbon radicals having from 1 to 12 carbon atoms such as, for example, 1,1-methanediyl, 1,2-ethanediyl, 1,3-propanediyl, 1,4-butanediyl, 1,5-pentanediyl, 1 ,6-hexanediyl, 1,2-propanediyl, 2,3-butanediyl, 1 ,7-heptanediyl, 1 ,8-octanediyl, 1,9-nonanediyl, 1,10-decan
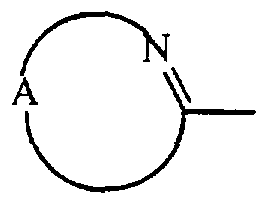
- the unsaturated heteroaryl group represented by ⁇ et may be attached to the remainder of the molecule of formula (I) through any ring carbon or heteroatom as appropriate
- the heteroaryl group when it is imidazolyl, it may be a 1 - ⁇ m ⁇ dazolyl, 2- ⁇ m ⁇ dazolyl, 4- ⁇ midazolyl and 5- ⁇ m ⁇ dazolyl; when it is t ⁇ azolyl, it may be 1 ,2,4-t ⁇ azol-l-yl, l ,2,4-t ⁇ azol-3-yl, l,2,4-t ⁇ azol-5-yl, 1 ,3,4-t ⁇ azol- l -yI and 1,3,4- t ⁇ azol-2-yl
- the pharmaceutically acceptable addition salts as mentioned hereinabove are meant to comprise the therapeutically active non-toxic base and acid addition salt forms which the compounds of formula (I) are able to form.
- the acid addition salt form of a compound of formula (I) that occurs in its free form as a base can be obtained by treating said free base form with an appropriate acid such as an inorganic acid, for example, hydrohalic acid, e.g.
- an organic acid such as, for example, acetic, hydroxyacetic, propanoic, lactic, pyruvic, oxalic, malonic, succmic, maleic, fumaric, malic, tarta ⁇ c, citric, methanesulfo c, ethanesulfomc, benzenesulfonic, -tolu
- the compounds of formula (I) containing acidic protons may be converted into their therapeutically active non-toxic base, i.e. metal or amine, addition salt forms by treatment with appropriate organic and inorganic bases.
- Appropriate base salt forms comprise, for example, the ammonium salts, the alkali and earth alkaline metal salts, e.g. the lithium, sodium, potassium, magnesium, calcium salts and the like, salts with organic bases, e.g. the benzathine, N-methyl-D-glucamine, hydrabamine salts, and salts with amino acids such as, for example, arginine, lysine and the like.
- salt forms can be converted into the free forms by treatment with an appropriate base or acid
- addition salt as used hereinabove also comprises the solvates which the compounds of formula (I) as well as the salts thereof, are able to form.
- solvates are for example hydrates, alcoholates and the like.
- N-oxide forms of the compounds of formula (I) are meant to comprise those compounds of formula (I) wherein one or several nitrogen atoms are oxidized to the so-called N-oxide.
- stereochemically isomeric forms as used hereinbefore and hereinafter defines all the possible isomeric forms in which the compounds of formula (I) may occur. Unless otherwise mentioned or indicated, the chemical designation of compounds denotes the mixture, and in particular the racemic mixture, of all possible stereochemically isomeric forms, said mixtures containing all diastereomers and enantiomers of the basic molecular structure. Stereochemically isomeric forms of the compounds of formula (I) and mixtures of such forms are obviously intended to be encompassed by formula (I).
- the term compounds of formula (I) is meant to include also the N-oxides, the pharmaceutically acceptable addition salts and all stereoisomeric forms.
- a particular group of compounds comprises those compounds of formula (I) wherein Ri represents hydrogen, Chalky! or aryl; R2 represents hydrogen; C-.i ⁇ alkyl; C 3 .7cycloalkyl; C2-8 l enyl; aryl; or
- R2 represents hydrogen; Ci. ⁇ alkyl, C 3 . 7 cycloalkyl; pyrrolidinyl optionally substituted with C ⁇ . 4 alkyl or C
- R3 represents hydrogen and C- ⁇ alkyl
- Het represents imidazolyl optionally substituted with Chalky I; pyridinyl or triazolyl, formyl or C- ⁇ alkyloxycarbonyl; 2-qu ⁇ noxahnyl; 1 -isoquinolmyl, 2-qu ⁇ nohnyl,
- R and R ⁇ each independently represent hydrogen, hydroxy, nitro, cyano, amino, C ⁇ _6alkyl or aryl; -R 6 -R 7 - represents a bivalent radical of formula (b- 1 ), (b-2) or (b- 10), wherein each R 9 independently represents hydrogen, C- ⁇ alkyl, hydroxy, halo, amino, haloC ⁇ .6alkyl or C ⁇ -0 alkyloxy
- Het is optionally substituted imidazolyl or triazolyl, in particular, 1 -imidazolyl optionally substituted with C- ⁇ alkyl or aryl, 2- ⁇ m ⁇ dazolyl optionally substituted with Ci- 0 alkyl; 5- ⁇ m ⁇ dazolyl optionally substituted with C-.ealkyl; 1 ,3,4-t ⁇ azol-l -yl and 1,2,4-t ⁇ azol-l-yl
- R ⁇ represents C substituted with mono- or d ⁇ (C ⁇ . 4 alkyl)am ⁇ no, C- ⁇ alkyloxycarbonyl or aryloxy
- Particular compounds are those compounds of special interest wherein Het is 1 -imidazolyl optionally substituted with Chalky! or aryl, 2- ⁇ m ⁇ dazolyl optionally substituted with C ⁇ alkyl, 5- ⁇ m ⁇ dazolyl optionally substituted with Ci ⁇ lkyl, 1 ,3,4-t ⁇ azol- l -yl and 1 ,2,4-t ⁇ azol- l-yl, R2 represents Ci
- Preferred compounds are those compounds of formula (I) wherein R 1 is hydrogen and R 2 is C3-7cycloalkyl or C-.6alkyl optionally substituted with d ⁇ (C ⁇ .6alkyl)am ⁇ no
- N-[4-[2-ethy I- 1 -( 1 H-imidazol- 1 -yl)butyl]phenyl]-2-benzothiazolamme N-[4-[2-ethy 1- 1 -( 1 H- 1 ,2,4-t ⁇ azol- 1 -yl)butyl]pheny l]-2-benzoxazolam ⁇ ne
- N-[4-[2-(dimethylamino)-l-(lH-imidazol-l -yl)propyl]phenyl]-2-benzothiazolamine N-[4-[2-(dimethylamino)- 1 -( 1H- 1 ,2,4-triazol- 1 -yl)
- R 1 to R 3 ⁇ et, aryl and are defined as under formula (I) unless otherwise indicated.
- the compounds of formula (I) can be prepared by reacting an intermediate of formula (II) wherein W 1 is an appropriate leaving group such as, for example, a halogen, hydroxy or an alkylsulfonyloxy group, with an intermediate of formula (III) or a functional derivative thereof.
- W 1 is an appropriate leaving group such as, for example, a halogen, hydroxy or an alkylsulfonyloxy group
- a functional derivative of imidazole may be l ,l '-carbonyldiimidazole.
- Said reaction may be performed in a reaction-inert solvent such as, for example, acetonitrile or tetrahydrofuran, in the presence of a suitable base such as, for example, potassium carbonate.
- a reaction-inert solvent such as, for example, acetonitrile or tetrahydrofuran
- a suitable base such as, for example, potassium carbonate.
- W 1 is an hydroxy group
- reaction products may be isolated from the reaction medium and, if necessary, further purified according to methodologies generally known in the art such as, for example, extraction, crystallization, distillation, trituration and chromatography.
- compounds of formula (I) may be prepared by N-alkylation of an intermediate of formula (IV) with an intermediate of formula (V) wherein W 2 is an appropriate leaving group such as, for example, a phenoxy group, in a reaction-inert solvent such as , for example, /V,N-dimethylformamide.
- intermediates of formula (VII) may be replaced by a functional derivative thereof such as, for example, the ketalized derivative thereof.
- a functional derivative thereof such as, for example, the ketalized derivative thereof.
- the reaction is suitably performed in the presence of an acid such as, for example, hydrochloric acid.
- the compounds of formula (I) wherein R 3 is hydrogen and is a radical of formula (b) wherein X represents S, said compounds being represented by formula (I-b-1), can be prepared by reacting an intermediate of formula (VIII) with an intermediate of formula (IX- 1) in a reaction-inert solvent such as, for example, tetrahydrofuran or l-methyl-2-pyrrolidinone.
- a reaction-inert solvent such as, for example, tetrahydrofuran or l-methyl-2-pyrrolidinone.
- intermediate (IX- 1 ) (VIII) (I-b-1 )
- intermediate (IX- 1 ) may be replaced by an intermediate of formula (IX-2) thus forming a compound of formula (I-a- 1 ) wherein R 3 is hydrogen and R 4 is amino, said compounds being represented by formula (I-a-2).
- reaction may also be performed using an intermediate of formula (X). Said reaction is then performed in a reaction-inert solvent such as, for example, dimethylsulfoxide, and in the presence of a suitable base such as, for example, sodium hydroxide.
- a reaction-inert solvent such as, for example, dimethylsulfoxide
- Compounds of formula (I) wherein R 2 is optionally substituted d. ⁇ alkyl can be prepared by reducing an intermediate corresponding to a compound of formula (I) wherein said R" is connected to the carbon atom bearing the R" substituent by a double bond using a suitable reducing agent such as, for example, sodiumborohydride, in a suitable solvent such as methanol.
- a suitable reducing agent such as, for example, sodiumborohydride
- the compounds of formula (I) can also be converted into each other following art-known procedures of functional group transformation.
- compounds of formula (I) wherein R 3 is hydrogen may be converted to compounds of formula (I) wherein R 3 is other than hydrogen.
- organic peroxides may comprise peroxy acids such as, for example, benzenecarboperoxoic acid or halo substituted benzenecarboperoxoic acid, e.g. 3-chlorobenzenecarboperoxoic acid, peroxoalkanoic acids, e.g. peroxoacetic acid, alkylhydroperoxides, e.g. t-butyl hydroperoxide.
- Suitable solvents are, for example, water, lower alkanols, e.g. ethanol and the like, hydro ⁇ carbons, e.g. toluene, ketones, e.g. 2-buta ⁇ one, halogenated hydrocarbons, e.g. dichloromethane, and mixtures of such solvents.
- the first reaction involves the same procedure as the one used hereinabove for the preparation of compounds of formula (I-b- 1 ) starting from an intermediate of formula (IX) and an intermediate of formula (VIII) or (X).
- the reduction may be performed in the presence of a suitable reducing agent in an appropriate reaction-inert solvent such as, for example, sodiumborohydride in methanol or lithiumaluminiumhydride in tetrahydrofuran and water.
- intermediates of formula (II-b-1) it may be convenient to replace the hydroxy group in intermediates of formula (II-b-1) by another leaving group such as, for example, a halogen or a sulfonyl derivative, e.g. a /?-toluenesulfonyloxy group or a alkylsulfonyloxy group, thus forming intermediates of formula (II-b-2) or (II-b-3).
- Said reaction can be performed in a reaction-inert solvent, such as, for example, chloroform, and in the presence of a suitable reagent such as, for example, thionylchloride or methylsulfonyl chloride.
- Intermediates of formula (IV) may be prepared by reacting an intermediate of formula (XII), wherein P is a protective group such as, for example, Ci ⁇ alkylcarbonyl, benzoyl or with an intermediate of formula (III), and by subsequently reacting the thus formed amide derivative with an acid such as, for example, hydro ⁇ chloric acid.
- the preparation of the intermediate amide derivative may be performed using the same procedure as the one used for the preparation of compounds of formula (I) starting form an intermediate of formula (II) and (III).
- Intermediates of formula (IV) wherein R 3 is hydrogen, said intermediates being represented by formula (IV-a), may also be reacted with an appropriate reagent such as CSCI 2 or a functional derivative thereof, in a reaction inert solvent and in the presence of a suitable base such as, for example, sodium hydroxide, thus forming intermediates of formula (V ⁇ i).
- an appropriate reagent such as CSCI 2 or a functional derivative thereof
- a suitable base such as, for example, sodium hydroxide
- intermediates of formula (IV-a) may further be used in the preparation of intermediates of formula (X).
- Said preparation involves the reaction of an intermediate of (IV-a) with CS2 and CH3-I or a functional derivatives of any one of said reagents, in a reaction-inert solvent and in the presence of a base such as, for example, sodium hydroxide.
- the compounds of formula (I) suppress the plasma elimination of retinoids, such as all- tra-w-retinoic acid, 13-cis retinoic acid and their derivatives, resulting in more sustained plasma and tissue concentrations of retinoic acid and improved control of the differentiation and growth of various cell types.
- This action of the present compounds is also called retinoic mimetic activity because administering a compound of formula (I) causes the same effect as if retinoids were administered.
- the present compounds can be used to control the rate of growth and differentiation of normal, preneoplastic and neoplastic cells, whether they are epithelial or mesenchymal; whether they are of ectodermal, endodermal or mesodermal origin.
- the property to delay the metabolism of retinoic acid can be evidenced in various in vitro and in vivo experiments.
- a particular in vitro procedure is described in example Cl and tests the inhibitory activity of the compounds of formula (I) on the metabolism of retinoic acid in human breast cancer cells.
- the compounds of the present invention were also effective in suppressing induced vaginal keratinization effects in ovariectomized rats as is described in example C.2.
- the compounds of formula (I) show little or no endocrinological side- effects and they have good oral availability.
- the present compounds are useful in the treatment and/or the prevention of disorders characterized by abnormal proliferation and/or abnormal differentiation of cells, in particular of cells of which the growth and differentiation is sensitive to the actions of retinoids.
- Such disorders are situated in the field of oncology, for example, head- and neck cancer, lung cancer, breast cancer, uterine cervix cancer, gastrointestinal tract cancer, skin cancer, bladder cancer and prostate cancer and similar disorders; and in the field of dermatology, for example, keratinization disorders such as rosacea, acne, psoriasis, severe psoriasis, lamellar ichthyosis, plantar warts, callosities, acanthosis nigricans, lichen planus, molluscum, melasma, corneal epithelial abrasion, geographic tongue, Fox-Fordyce disease, cutaneous metastatic melanoma and keloids, epidermolytic hyperkeratosis, Darier's disease, pityriasis rubra pilaris, congenital ichthyosiform erythroderma, hyperkeratosis palma ⁇ s et plantaris, melas
- the compounds of formula (I) are useful in suppressing the metabolism of exogenously administered and of endogenously formed 1 ⁇ ,25-dihydroxy-vitamin D 3
- the inhibitory activity of the compounds of formula (I) on the metabolic degradation of calcitriol may be evidenced by measuring the impact of said compounds on the calcitriol degradation in human foreskin keratinocytes, pig kidney cells and human hepatoma cells.
- the compounds of formula (I) can be used in the treatment of vitamin D deficiency states.
- the "classic" application of vitamin D compounds lies in the field of metabolic bone disorders. Calcitriol has also been described to influence the effects and/or production of interleukins.
- calcitriol is of use in the treatment of diseases characterized by abnormal cell proliferation and/or differentiation, in particular, keratinization disorders such as those described hereinabove (Bouillon et al.. Endocrine Reviews, 1995, 76, 200-257).
- the present invention provides a method of treating warm-blooded animals suffering from diseases which are characterized by an abnormal proliferation and/or abnormal differentiation of normal, preneoplastic or neoplastic cells, whether they are epithelial or mesenchymal; whether they are of ectodermal, endodermal or mesodermal origin.
- Said method comprises the systemic or topical administration of a retinoic mimetic amount of a compound of formula (I) effective in treating the above described disorders, in particular keratinization disorders such as psoriasis, optionally in the presence of an effective amount of a retinoic acid, a derivative or a stereochemically isomeric form thereof.
- the present invention further concerns a method of treating patients suffering from a pathological condition which may be beneficially influenced by the administration of calcitriol or a prodrug thereof, in particular keratinization disorders such as psoriasis, said method consisting of administering to a patient (a) an effective amount of calcitriol or a prodrug thereof and (b) an effective amount of a compound of formula (I).
- the present invention also relates to compounds of formula (I) as defined hereinabove for use as a medicine, in particular, for use in the manufacture of a medicament for the treatment of keratinization disorders such as psoriasis.
- the present invention further relates to compounds of formula (I) as defined hereinabove in combination with a retinoic acid, a derivative or a stereochemically isomeric form thereof, or in combination with calcitriol or a prodrug thereof, for use as a medicine.
- the subject compounds may be formulated into various pharmaceutical forms.
- compositions there may be cited all compositions usually employed for systemically or topically administering drugs.
- a retinoic mimetic effective amount of the particular compound, optionally in addition salt form, as the active ingredient is combined in intimate admixture with a pharmaceutically acceptable carrier, which may take a wide variety of forms depending on the form of preparation desired for administration.
- a pharmaceutically acceptable carrier which may take a wide variety of forms depending on the form of preparation desired for administration.
- These pharmaceutical compositions are desirably in unitary dosage form suitable, preferably, for administration orally, rectally, percutaneously, or by parenteral injection.
- any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols and the like in the case of oral liquid preparations such as suspensions, syrups, elixirs and solutions; or solid carriers such as starches, sugars, kaolin, lubricants, binders, disintegrating agents and the like in the case of powders, pills, capsules and tablets. Because of their ease in administration, tablets and capsules represent the most advantageous oral dosage unit form, in which case solid pharmaceutical carriers are obviously employed.
- the carrier will usually comprise sterile water, at least in large part, though other mgredients, for example, to aid solubility, may be included
- Injectable solutions for example, may be prepared in which the carrier comprises saline solution, glucose solution or a mixture of saline and glucose solution.
- the carrier optionally comprises a penetration enhancm**- agent and/or a suitable wettable agent, optionally combined with suitable additives of any nature in minor proportions, which additives do not cause any significant deleterious effects on the skin.
- compositions may be administered in various ways, e.g as a transdermal patch, as a spot-on or as an ointment.
- Addition salts of compounds of formula (I) due to their increased water solubility over the corresponding base form, are obviously more suitable in the preparation of aqueous compositions
- compositions for topical application there may be cited all compositions usually employed for topically administering drugs e.g creams, gellies, dressings, shampoos, tinctures, pastes, ointments, salves, powders and the like
- Application of said compositions may be by aerosol, e.g with a propellent such as nitrogen, carbon dioxide, a freon, or without a propellent such as a pump spray, drops, lotions, or a semisohd such as a thickened composition which can be applied by a swab
- a propellent such as nitrogen, carbon dioxide, a freon
- a propellent such as a pump spray
- drops lotions
- a semisohd such as a thickened composition which can be applied by a swab
- semisohd compositions such as salves, creams, gellies, ointments and the like will conveniently be used.
- Dosage unit form refers to physically discrete units suitable as unitary dosages, each unit containing a predetermined quantity of active ingredient calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier.
- dosage unit forms are tablets (included scored or coated tablets), capsules, pills, powder packets, wafers, injectable solutions or suspensions, teaspoonfuls, tablespoonfuls and the like, and segregated multiples thereof.
- compositions of the cosmetic type, such as toilet waters, packs, lotions, skin milks or milky lotions
- Said preparations contain, besides the active ingredient, components usually employed in such preparations
- components usually employed in such preparations
- further ingredients may be inco orated in the compositions, e.g. antiinflamatory agents, antibacterials, antifungals, disinfectants, vitamins, sunscreens, antibiotics, or other anti-acne agents.
- the present invention also provides particular pharmaceutical or cosmetical compositions which comprise an inert carrier, an effective amount of a compound of formula (I) and an effective amount of a retinoic acid, a derivative thereof or a stereo ⁇ chemically isomeric form thereof.
- Said retinoic acid containing compositions are particularly useful for treating acne or for retarding the effects of aging of the skin and generally improve the quality of the skin, particularly human facial skin.
- the invention also relates to particular pharmaceutical or cosmetical compositions which comprise an inert carrier, an effective amount of a compound of formula (I) and an effective amount of calcitriol or a prodrug thereof. The latter compositions are particularly useful in treating keratinization disorders.
- the invention also relates to a product containing retinoic acid or a derivative thereof and a compound of formula (I) as a combined preparation for simultaneous, separate or sequential use in dermatological or oncological disorders.
- the invention also relates to a product containing calcitriol or a prodrug thereof and a compound of formula (I) as a combined preparation for simultaneous, separate or sequential use in disorders benefi- cially affected by calcitriol.
- Such products may comprise, for example, a kit comprising a container with a suitable composition containing a compound of formula (I) and another container with a composition containing calcitriol or a retinoid
- a kit comprising a container with a suitable composition containing a compound of formula (I) and another container with a composition containing calcitriol or a retinoid
- Such a product may have the advantage that a physician can select on the basis of the diagnosis of the patient to be treated the appropriate amounts of each component and the sequence and timing of the administration thereof.
- an effective therapeutic daily amount would be from about 0.01 mg/kg to about 40 mg/kg body weight, more preferably from about 0.1 mg/kg to about 10 mg/kg body weight. It may be appropriate to administer the therapeutically effective dose once daily or as two, three, four or more sub-doses at appropriate intervals throughout the day. Said sub-doses may be formulated as unit dosage forms, for example, containing 0.1 mg to 500 mg of active ingredient per unit dosage form.
- the exact dosage and frequency of administration depends on the particular compound of formula (I) used, the particular condition being treated, the severity of the condition being treated, the age, weight and general physical condition of the particular patient as well as other medication the patient may be taking, as is well known to those skilled in the art. Furthermore, it is evident that said effective daily amount may be lowered or increased depending on the response of the treated patient and/or depending on the evaluation of the physician prescribing the compounds of the instant invention. The effective daily amount ranges mentioned hereinabove are therefore only guidelines.
- THF tetrahydrofuran
- EtOAc ethylacetate
- DIPE diisopropyl ether
- RT room temperature
- Example A- 1 A) Preparation of the intermediate compounds Example A- 1 a) Benzoyl chloride (0.067 mol) was added to a solution of aminothiocyanate (5.09g) in 2 propanone (150 ml) and the mixture was stirred and refluxed for 20 minutes. A solution of 4-[l -(lH-imidazol-l-yl)-2-methylpropyl]benzenamine (0.0557 mol) in 2-propanone ( 150 ml) was added and the mixture was stirred and refluxed at 80°C overnight. The mixture was cooled, filtered through celite and the filtrate was evaporated. The residue was taken up in C ⁇ 2CI2. The organic layer was dried, filtered off and the solvent evaporated.
- Example A-2 a) Sec butyllithium (298 ml; 1.3 M) was added dropwise at -60°C under N2 flow to a solution of N-(4-bromophenyl)acetamide (0.1892 mol) in T ⁇ F (400 ml) and the mixture was stirred at -70°C for 2 hours. A solution of 1-cyano-l -methyl -NN-di- methylethanamme (0.075 mol) in T ⁇ F (60 ml) was added dropwise, the mixture was brought to RT and then stirred at RT for 12 hours. The mixture was poured into ice and extracted with EtOAc.
- Example A-3 a) A solution of lithium tetrahydroaluminate (0.1 107 mol) in THF (100 ml) was added dropwise at 0°C under N2 flow to a suspension of ethyl 4-(2-benzothiazolylamino)- benzoate (0.1 107 mol) in water. The mixture was brought to RT and stirred for 30 minutes. The mixture was hydrolized by adding water (8 ml) dropwise and then CH2CI2 (50 ml), and a little CH3OH was added. The precipitate was filtered and the solvent was evaporated. The residue was crystallized from 2-propanone and DIPE.
- Example A-4 a) A mixture of intermediate (8) (0.0312 mol) and manganese dioxide (0.1 15 mol) in CH2CI2 (200ml) and NN-dimethylformamide ( 10 ml) was stirred at to RT for 12 hours. Manganese dioxide (0.1 15 mol) was added again and the mixture was stirred at RT for 12 hours. The mixture was filtered through celite, washed with CH2CI2 and the solvent was evaporated. Water (100 ml) was added, evaporated, filtered, crystallized, filtered and dried, yielding 7 g (89%) of 4-(2-benzothiazolylamino)benzaldehyde (interm. 10). b) A solution of 1 -bromo-3-fluorobenzene (0.213 mol) in THF (60 ml) was added dropwise at RT under ⁇ 2 flow to a suspension of magnesium (0.213 mol) in THF
- Example A-5 a) A solution of 3-bromopentane (0.331 mol) in (C Hs)2 ⁇ (200ml) was added dropwise to a solution of magnesium turnings (0.331 mol) in (C 2 Hs)2O, the mixture was stirred at RT for 2 hours and then cooled to 0°C. A solution of /V-(4-formyl- phenyl)acetamide (0.1 1 mol) in THF (400 ml) was added dropwise and the mixture was stirred for 10 minutes. The mixture was poured into aqueous NH4CI and extracted with
- Example A-6 a) A mixture of 1 -(4-aminophenyl)-2-methyl- 1 -propanone (0.0637 mol) and methyl 2-chloro-3-pyridinecarboxylate (0.0637 mol) in 2-methoxyethanol (200 ml) was stirred and refluxed for 90 hours. The mixture was taken up in water and EtOAc and extracted with EtOAc. The organic layer was separated, dried, filtered and the solvent was evaporated, yielding 22.6 g of methyl 2-[[4-(2-methyl- l-oxopropyl)phenyl]amino]-3- pyridinecarboxylate (inte ⁇ n. 20).
- Example A-7 a) Aluminium(III)chloride (0.666 mol) was added portionwise at RT to a solution of N- ⁇ henyl-2-benzothiazolylamine (0.222 mol) and 1 ,2-dichloro- 1 -propanone (0.233 mol) in 1,2-dichloroethane (500 ml) and the mixture was stirred and heated at 80°C for 2 hours. The mixture was poured into ice and extracted with CH2CI2. The organic layer was decanted, dried filtered and the solvent was evaporated, yielding 68 g of ( ⁇ )-l-[4-(2-benzothiazolylamino)phenyl]-2-chloro- 1 -propanone (95.7%) (interm. 22).
- Example A.8 a The following reaction was performed under a N atmosphere. A mixture of 7V-(4- bromophenyl)-2-benzothiazolarnine (0.492 mol) in THF (2700 ml) was stirred at -70°C. Butyllithium (0.984 mol; 2.5 M in hexane) was added dropwise at -65 °C. The mixture was stirred for one hour. A solution of 2-ethyl-butanal (0.492 mol) in THF (300 ml) was added dropwise at -75 °C. The mixture was allowed to warm to RT overnight. A 10% aqueous NH C1 solution (3000 ml) was added and the mixture was stirred for 15 minutes.
- Example B-2 Tripheny Iphosphine (4.8g) and 1 H- 1 ,2,4-triazole (0.018 mol) were added under N2 flow at 5°C to a solution of intermediate (7) (0.00732 mol) in T ⁇ F. Then a solution of diethyl azodicarboxylate (2.88 ml) in T ⁇ F was added, the mixture was brought to RT and then stirred for overnight. Water was added, the solvent was evaporated, acidified with HC1 (3 N) and the layers were separated. The aqueous layer was washed with EtOAc, basified with NH4OH and extracted with EtOAc. The organic layer was separated, dried, filtered and the solvent was evaporated. The residue was purified by column chromatography over silica gel (eluent : CH2CI2/CH3OH NH4OH 96/4/0.5).
- Example B-5 a) 1, 1 '-carbonylbis- IH-imidazole (0.122 mol) was added at 60°C to a mixture of intermediate (21 ) (0.0612 mol) in T ⁇ F (250 ml). The mixture was stirred overnight, poured out into water and extracted with EtOAc. The organic layer was separated, dried, filtered and the solvent was evaporated. The residue was purified by column chromatography over silica gel (eluent: C ⁇ 2CI2/C ⁇ 3O ⁇ /N ⁇ 4O ⁇ 99.25/0.75/0.1 ).
- Example B-8 A mixture of intermediate (2) (0.0269 mol) and 2-bromo- 1 , 1 -diethoxyethane (0.035 mol) in ⁇ C1 (1 1.8 ml; 3 ⁇ ) and ethanol (200 ml) was stirred and refluxed for 3 hours. The mixture was cooled and evaporated. The residue was taken up in C ⁇ 2CI2 and K2CO3 (10%) and extracted with CH2CI2. The organic layer was washed with water and K2CO3 ( 10%), dried, filtered off and the solvent evaporated. The residue was purified by column chromatography over silica gel (eluent : CH2CI2/CH3OH/ ⁇ H4OH 96/4/0.2). The pure fractions were collected and evaporated.
- Example B- 10 A mixture of intermediate 43 (0.0089 mol) and CH ONa 30% in CH 3 OH (0.0445 mol) in CH 3 OH (81ml) was stirred and refluxed for 15 hours. The mixture was cooled, poured out into water, saturated with NaCl and extracted with CH C1 2 . The organic layer was separated, dried, filtered and the solvent was evaporated. The residue (3.3g) was purified by column chromatography over silica gel (eluent : CH 2 Cl 2 /CH 3 OH NH 4 OH 95/5/0.2; 15-40 ⁇ m). Two pure fractions were collected and their solvents were evaporated. Fraction 1 was crystallized from 2-butanone and diethyl ether.
- Example B- 14 A mixture of intermediate (26) (0.156 mol), lH-l ,2,4-triazole (0.313 mol) and K CO 3 (0.313 mol) in C ⁇ 3 C ⁇ (800ml) was stirred and refluxed for 12 hours. The solvent was evaporated. The residue was taken up in CH 2 Cl /H 2 O. The organic layer was separated, dried, filtered and the solvent was evaporated. The residue was purified by column chromatography over silica gel (eluent: CH 2 Cl 2 /CH 3 OH/NH OH 98/2/0.1). The pure fractions were collected and the solvent was evaporated. The residue was crystallized from diethyl ether.
- Example B- 15 n-Butyl lithium ( 1.6 M; 0.0607 mol) was added dropwise at -70°C under ⁇ 2 flow to a solution of 1 -methyl- IH-imidazole (0.0607 mol) in T ⁇ F (60ml). The mixture was stirred at -70°C for 30 minutes. A mixture of intermediate 24 (0.0243 mol) in T ⁇ F (60ml) was added dropwise. The mixture was stirred and poured out into water and N ⁇ CI. The organic layer was separated, dried, filtered and the solvent was evaporated. The residue was purified by column chromatography over silica gel (eluent: CH Cl 2 /CH 3 OH NH 4 OH 98/2/0.2).
- Tables 1 to 6 list compounds of formula (I) that were prepared according to one of the above examples.
- Table 7 lists both the experimental (column heading “exp”) and theoretical (column heading “theor”) elemental analysis values for carbon, hydrogen and nitrogen of the compounds as prepared in the experimental part hereinabove.
- Example C.1 Inhibition of retinoic acid (RA) metabolism
- MCF-7 human breast cancer cells were grown as stock cultures according to art-known protocols.
- RA is added to the stock cultures to stimulate RA-metabolism.
- cell suspensions were incubated in a tissue culture medium containing 3 H-RA as the substrate.
- Different concentrations of the test compound dissolved in 1% DMSO
- the unmetabolized RA is separated from its polar metabolites.
- the fraction containing the polar 3 H-labelled metabolites was collected and counted in a scintillation counter.
- a control and a blank incubation were run in parallel. Those compounds that were tested, i.e.
- estradiol undecylate 100 ⁇ g of estradiol undecylate in a volume of 0.1 ml per 100 g body weight and control animals were injected with sesame oil.
- test animals were treated once daily with a per os dose of the test compound and control animals with the drug vehicle (PEG 200).
- PEG 200 drug vehicle
- the animals were sacrificed and their vaginas were processed for histological evaluation according to the method described in J. Pharmacol. Exp. Ther. 267(2), 773-779 ( 1992).
- a dose at which 50 % of the tested rats show complete suppression of the estradiol undecylate induced keratinization effects is defined as an active dose.
- LAD active dose
- Active ingredient as used throughout these examples relates to a compound of formula (I) or a pharmaceutically acceptable acid addition salt thereof.
- Example D.1 oral solution
- Example D.3 capsules 20 g of A.I., 6 g sodium lauryl sulfate, 56 g starch, 56 g lactose, 0.8 g colloidal silicon dioxide, and 1.2 g magnesium stearate were vigorously stirred together. The resulting mixture was subsequently filled into 1000 suitable hardened gelatin capsules, each comprising 20 mg of A.I.
- Example D.5 film-coated tablets Preparation of tablet, core
- the wet powder mixture was sieved, dried and sieved again.
- the whole was mixed well and compressed into tablets, giving 10.000 tablets, each comprising 10 mg of the active ingredient.
- Example D.6 2% cream
- 75 mg stearyl alcohol, 2 mg cetyl alcohol, 20 mg sorbitan monostearate and 10 mg isopropyl myristate are introduced into a doublewall jacketed vessel and heated until the mixture has completely molten.
- This mixture is added to a separately prepared mixture of purified water, 200 mg propylene glycol and 15 mg polysorbate 60 having a temperature of 70 to 75°C while using a homogenizer for liquids.
- the resulting emulsion is allowed to cool to below 25°C while continuously mixing.
- a solution of 20 mg A.I., 1 mg polysorbate 80 and purified water and a solution of 2 mg sodium sulfite anhydrous in purified water are next added to the emulsion while continuously mixing.
- the cream, 1 g of the A.I. is homogenized and filled into suitable tubes.
- Example D.7 2% topical gel
- Example D.8 2% topical cream
- a mixture of 2 g A.I. microfine, 20 g phosphatidyl choline, 5 g cholesterol and 10 g ethyl alcohol is stirred and heated at 55-60°C until complete dissolution and is added to a solution of 0.2 g methyl paraben. 0.02 g propyl paraben, 0.15 g disodium edetate and 0.3 g sodium chloride in purified water while homogenizing. 0.15 g Hydroxypropyl- methylcellulose in purified water ad 100 g is added and the mixing is continued until swelling is complete.
Abstract
Description





















































Claims




















Priority Applications (21)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE69717627T DE69717627T2 (en) | 1996-06-27 | 1997-06-19 | N-4- (heteroarylmethyl) phenyl-HETEROARYLAMINDERIVATE |
| IL12774097A IL127740A (en) | 1996-06-27 | 1997-06-19 | N-[4-(heteroarylmethyl) phenyl] -heteroarylamines and their n-oxides, their preparation, and pharmaceutical compositions comprising them |
| US09/214,080 US6124330A (en) | 1996-06-27 | 1997-06-19 | N-[4-(Heteroarylmethyl)phenyl]-heteroarylamines |
| NZ333382A NZ333382A (en) | 1996-06-27 | 1997-06-19 | N-[4-(heteroarylmethyl)phenyl]-heteroarylamines |
| SI9730478T SI0907650T1 (en) | 1996-06-27 | 1997-06-19 | N- 4-(heteroarylmethyl)phenyl)-heteroarylamines |
| JP50232198A JP3404749B2 (en) | 1996-06-27 | 1997-06-19 | N- [4- (heteroarylmethyl) phenyl] -heteroarylamine |
| EE9800437A EE03688B1 (en) | 1996-06-27 | 1997-06-19 | N- [4- (Heteroarylmethyl) phenyl] -heteroarylamines, their Preparation and Use, Pharmaceutical Composition and Method for its Preparation |
| EP97930378A EP0907650B1 (en) | 1996-06-27 | 1997-06-19 | N- 4-(heteroarylmethyl)phenyl]-heteroarylamines |
| HU9902138A HU223093B1 (en) | 1996-06-27 | 1997-06-19 | N-[4-(heteroarylmethyl)phenyl]-heteroarylamine derivatives, their preparation and pharmaceutical compositions containing them |
| UA98116146A UA53649C2 (en) | 1996-06-27 | 1997-06-19 | N-[4-(heteroarylmethyl)phenyl]heteroarylamines, composition on basis thereof and a process for preparation thereof, a process for compounds preparation (variants) |
| DK97930378T DK0907650T3 (en) | 1996-06-27 | 1997-06-19 | N-4- (heteroarylmethyl) phenyl] heteroaryl amines |
| CA002258165A CA2258165C (en) | 1996-06-27 | 1997-06-19 | N-[4-(heteroarylmethyl)phenyl]-heteroarylamines |
| BR9710002A BR9710002A (en) | 1996-06-27 | 1997-06-19 | N- [4- (heteroarylmethyl) phenyl] -heteroaryl amines |
| AT97930378T ATE229019T1 (en) | 1996-06-27 | 1997-06-19 | N-4-(HETEROARYLMETHYL)PHENYL-HETEROARYLAMINE DERIVATIVES |
| SK1781-98A SK282769B6 (en) | 1996-06-27 | 1997-06-19 | N-[4-(Heteroarylmethyl)phenyl]-heteroarylamines |
| AU34356/97A AU711575B2 (en) | 1996-06-27 | 1997-06-19 | N-(4-(Heteroarylmethyl)Phenyl)-Heteroarylamines |
| BG103013A BG63545B1 (en) | 1996-06-27 | 1998-12-14 | N-[4-(heteroarylmethyl)phenyl]-heteroarylamines |
| NO19986017A NO312102B1 (en) | 1996-06-27 | 1998-12-21 | N- [4- (heteroarylmethyl) phenyl] -Heteroarylaminer |
| HK99103546A HK1018458A1 (en) | 1996-06-27 | 1999-08-16 | N-4-(heteroarylmethyl)phenyl]-heteroarylamines |
| US10/979,362 US7205312B2 (en) | 1996-06-27 | 2004-11-02 | N-[4-(heteroarylmethyl)phenyl]-heteroarylamines |
| US11/639,043 US7378433B2 (en) | 1996-06-27 | 2006-12-13 | N-[4-(heteroarylmethyl)phenyl]-heteroarylamines |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP96201781.0 | 1996-06-27 | ||
| EP96201781 | 1996-06-27 |
Related Child Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/214,080 A-371-Of-International US6124330A (en) | 1996-06-27 | 1997-06-19 | N-[4-(Heteroarylmethyl)phenyl]-heteroarylamines |
| US09214080 A-371-Of-International | 1997-06-19 | ||
| US09/624,966 Division US6486187B1 (en) | 1996-06-27 | 2000-07-25 | N-[4-(heteroarylmethyl)phenyl]-heteroarylamines |
| US09/624,966 Continuation US6486187B1 (en) | 1996-06-27 | 2000-07-25 | N-[4-(heteroarylmethyl)phenyl]-heteroarylamines |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1997049704A1 true WO1997049704A1 (en) | 1997-12-31 |
Family
ID=8224119
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1997/003248 WO1997049704A1 (en) | 1996-06-27 | 1997-06-19 | N-[4-(heteroarylmethyl)phenyl]-heteroarylamines |
Country Status (33)
| Country | Link |
|---|---|
| US (5) | US6124330A (en) |
| EP (1) | EP0907650B1 (en) |
| JP (1) | JP3404749B2 (en) |
| KR (1) | KR100365312B1 (en) |
| CN (1) | CN1102593C (en) |
| AR (1) | AR007626A1 (en) |
| AT (1) | ATE229019T1 (en) |
| AU (1) | AU711575B2 (en) |
| BG (1) | BG63545B1 (en) |
| BR (1) | BR9710002A (en) |
| CA (1) | CA2258165C (en) |
| CZ (1) | CZ297769B6 (en) |
| DE (1) | DE69717627T2 (en) |
| DK (1) | DK0907650T3 (en) |
| EE (1) | EE03688B1 (en) |
| ES (1) | ES2188957T3 (en) |
| HK (1) | HK1018458A1 (en) |
| HU (1) | HU223093B1 (en) |
| ID (1) | ID17275A (en) |
| IL (1) | IL127740A (en) |
| MY (1) | MY116917A (en) |
| NO (1) | NO312102B1 (en) |
| NZ (1) | NZ333382A (en) |
| PL (1) | PL201704B1 (en) |
| PT (1) | PT907650E (en) |
| RU (1) | RU2190611C2 (en) |
| SI (1) | SI0907650T1 (en) |
| SK (1) | SK282769B6 (en) |
| TR (1) | TR199802709T2 (en) |
| TW (1) | TW490464B (en) |
| UA (1) | UA53649C2 (en) |
| WO (1) | WO1997049704A1 (en) |
| ZA (1) | ZA975698B (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999029674A1 (en) * | 1997-12-11 | 1999-06-17 | Janssen Pharmaceutica N.V. | Retinoic acid mimetic anilides |
| US6291502B1 (en) | 1999-10-27 | 2001-09-18 | Aventis Pharma Deutschland Gmbh | Use of 2-imidazolyl-substituted carbinols for the production of a medicament for the treatment or phophylaxis of diseases caused by ischemic conditions |
| US6833373B1 (en) | 1998-12-23 | 2004-12-21 | G.D. Searle & Co. | Method of using an integrin antagonist and one or more antineoplastic agents as a combination therapy in the treatment of neoplasia |
| WO2007041379A1 (en) * | 2005-09-30 | 2007-04-12 | Bristol-Myers Squibb Company | Met kinase inhibitors |
| US7399764B2 (en) | 2002-10-30 | 2008-07-15 | Merck & Co., Inc. | Inhibitors of Akt activity |
| US7638530B2 (en) | 2003-04-24 | 2009-12-29 | Merck & Co., Inc. | Inhibitors of Akt activity |
| US7989462B2 (en) | 2003-07-03 | 2011-08-02 | Myrexis, Inc. | 4-arylamin-or-4-heteroarylamino-quinazolines and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| US8258145B2 (en) | 2005-01-03 | 2012-09-04 | Myrexis, Inc. | Method of treating brain cancer |
| US8309562B2 (en) | 2003-07-03 | 2012-11-13 | Myrexis, Inc. | Compounds and therapeutical use thereof |
| US8513291B2 (en) | 2010-06-01 | 2013-08-20 | Angion Biomedica Corp. | Cytochrome P450 inhibitors and uses thereof |
| CN106588779A (en) * | 2016-12-15 | 2017-04-26 | 青岛辰达生物科技有限公司 | Method for synthesizing dexmedetomidine hydrochloride intermediate |
| CN106588780A (en) * | 2016-12-15 | 2017-04-26 | 青岛辰达生物科技有限公司 | Process for preparing dexmedetomidine hydrochloride intermediate |
| CN106632051A (en) * | 2016-12-15 | 2017-05-10 | 青岛辰达生物科技有限公司 | Synthetic method of dexmedetomidine hydrochloride intermediate |
| US9988374B2 (en) | 2014-08-11 | 2018-06-05 | Angion Biomedica Corp. | Cytochrome P450 inhibitors and uses thereof |
| US10414760B2 (en) | 2016-11-29 | 2019-09-17 | Angion Biomedica Corp. | Cytochrome P450 inhibitors and uses thereof |
| US10604533B2 (en) | 2010-11-19 | 2020-03-31 | Ligand Pharmaceuticals Incorporated | Heterocycle amines and uses thereof |
| US10807983B2 (en) | 2015-03-16 | 2020-10-20 | Ligand Pharmaceuticals, Inc. | Imidazo-fused heterocycles and uses thereof |
| US11434234B2 (en) | 2014-12-31 | 2022-09-06 | Angion Biomedica Corp. | Methods and agents for treating disease |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR9710002A (en) * | 1996-06-27 | 1999-08-10 | Janssen Pharmaceutica Nv | N- [4- (heteroarylmethyl) phenyl] -heteroaryl amines |
| US7780687B2 (en) * | 2002-04-17 | 2010-08-24 | Tyco Healthcare Group Lp | Method and apparatus for anastomosis including expandable anchor |
| EP1660092A2 (en) | 2003-07-03 | 2006-05-31 | Myriad Genetics, Inc. | 4-arylamino-quinazolines as activators of caspases and inducers of apoptosis |
| DE10337942A1 (en) * | 2003-08-18 | 2005-03-17 | Merck Patent Gmbh | aminobenzimidazole derivatives |
| WO2006036497A2 (en) * | 2004-09-24 | 2006-04-06 | Allergan, Inc. | 4-(condensed cyclicmethyl)-imidazole-2-thiones acting as alpha2 adrenergic agonists |
| NZ553341A (en) | 2004-09-24 | 2010-11-26 | Allergan Inc | 4-(phenylmethyl and substituted phenylmethyl)-imidazole-2-thiones acting as specific alpha2 adrenergic agonists |
| BRPI0516025A (en) * | 2004-09-24 | 2008-08-19 | Allergan Sales Inc | 4- (heteroaryl-methyl and substituted heteroaryl-methyl) -imidazol-2-thione acting as alpha2-adrenergic agonists |
| CA2582071A1 (en) * | 2004-09-28 | 2006-04-06 | Allergan, Inc. | Unsubstituted and substituted 4-benzyl-1,3-dihydro-imidazole-2-thiones acting as specific or selective alpha2 adrenergic agonists and methods for using the same |
| AU2007207607B2 (en) * | 2006-01-17 | 2012-04-19 | Stiefel Laboratories, Inc. | Treatment of inflammatory disorders with triazole compounds |
| MX2009004096A (en) * | 2006-10-17 | 2009-06-16 | Stiefel Laboratories | Talarazole metabolites. |
| RU2475486C2 (en) * | 2007-10-18 | 2013-02-20 | Домпе С.П.А. | (r)-4-(heteroaryl)phenylehtyl derivatives and pharmaceutical compositions containing them |
| GB0811091D0 (en) * | 2008-06-17 | 2008-07-23 | Cancer Rec Tech Ltd | CYP26 Inhibitors |
| KR101108577B1 (en) * | 2010-07-28 | 2012-01-30 | 국제엘렉트릭코리아 주식회사 | Tape heater |
| US9248120B2 (en) | 2011-08-23 | 2016-02-02 | The Board Of Trustees Of The Leland Stanford Junior University | Reversing intestinal inflammation by inhibiting retinoic acid metabolism |
| KR20210055340A (en) | 2019-11-07 | 2021-05-17 | 정화종 | Luggage cover |
| CN115462347A (en) * | 2021-08-13 | 2022-12-13 | 广州市妇女儿童医疗中心 | Application of retinoic acid metabolic pathway in congenital megacolon diseases |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0260744A2 (en) * | 1986-09-15 | 1988-03-23 | Janssen Pharmaceutica N.V. | (1H-imidazol-1-ylmethyl) substituted benzimidazole derivatives |
| EP0371559A2 (en) * | 1988-11-29 | 1990-06-06 | Janssen Pharmaceutica N.V. | Use of benzimidazoles in the treatment of epithelial disorders |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU634655B2 (en) | 1990-04-25 | 1993-02-25 | Nissan Chemical Industries Ltd. | Pyridazinone derivative |
| TW321649B (en) | 1994-11-12 | 1997-12-01 | Zeneca Ltd | |
| US5582832A (en) * | 1995-06-06 | 1996-12-10 | Chesebrough-Pond's Usa Co., Division Of Conopco, Inc. | Compositions for topical application to skin |
| GB9624482D0 (en) | 1995-12-18 | 1997-01-15 | Zeneca Phaema S A | Chemical compounds |
| HUP9901155A3 (en) | 1996-02-13 | 2003-04-28 | Astrazeneca Ab | Quinazoline derivatives as vegf inhibitors |
| US5731342A (en) * | 1996-02-22 | 1998-03-24 | Eli Lilly And Company | Benzothiophenes, formulations containing same, and methods |
| BR9710002A (en) * | 1996-06-27 | 1999-08-10 | Janssen Pharmaceutica Nv | N- [4- (heteroarylmethyl) phenyl] -heteroaryl amines |
-
1997
- 1997-06-19 BR BR9710002A patent/BR9710002A/en not_active IP Right Cessation
- 1997-06-19 ES ES97930378T patent/ES2188957T3/en not_active Expired - Lifetime
- 1997-06-19 WO PCT/EP1997/003248 patent/WO1997049704A1/en active IP Right Grant
- 1997-06-19 HU HU9902138A patent/HU223093B1/en active IP Right Grant
- 1997-06-19 KR KR10-1998-0709764A patent/KR100365312B1/en not_active IP Right Cessation
- 1997-06-19 NZ NZ333382A patent/NZ333382A/en not_active IP Right Cessation
- 1997-06-19 UA UA98116146A patent/UA53649C2/en unknown
- 1997-06-19 DE DE69717627T patent/DE69717627T2/en not_active Expired - Lifetime
- 1997-06-19 IL IL12774097A patent/IL127740A/en not_active IP Right Cessation
- 1997-06-19 AU AU34356/97A patent/AU711575B2/en not_active Expired
- 1997-06-19 CA CA002258165A patent/CA2258165C/en not_active Expired - Lifetime
- 1997-06-19 SK SK1781-98A patent/SK282769B6/en not_active IP Right Cessation
- 1997-06-19 PL PL330816A patent/PL201704B1/en unknown
- 1997-06-19 CZ CZ0423198A patent/CZ297769B6/en not_active IP Right Cessation
- 1997-06-19 US US09/214,080 patent/US6124330A/en not_active Expired - Lifetime
- 1997-06-19 CN CN97195865A patent/CN1102593C/en not_active Expired - Lifetime
- 1997-06-19 PT PT97930378T patent/PT907650E/en unknown
- 1997-06-19 EP EP97930378A patent/EP0907650B1/en not_active Expired - Lifetime
- 1997-06-19 TR TR1998/02709T patent/TR199802709T2/en unknown
- 1997-06-19 AT AT97930378T patent/ATE229019T1/en active
- 1997-06-19 DK DK97930378T patent/DK0907650T3/en active
- 1997-06-19 EE EE9800437A patent/EE03688B1/en unknown
- 1997-06-19 JP JP50232198A patent/JP3404749B2/en not_active Expired - Lifetime
- 1997-06-19 RU RU99101902/04A patent/RU2190611C2/en active
- 1997-06-19 SI SI9730478T patent/SI0907650T1/en unknown
- 1997-06-23 TW TW086108726A patent/TW490464B/en not_active IP Right Cessation
- 1997-06-25 MY MYPI97002859A patent/MY116917A/en unknown
- 1997-06-26 ID IDP972217A patent/ID17275A/en unknown
- 1997-06-26 AR ARP970102822A patent/AR007626A1/en active IP Right Grant
- 1997-06-26 ZA ZA975698A patent/ZA975698B/en unknown
-
1998
- 1998-12-14 BG BG103013A patent/BG63545B1/en unknown
- 1998-12-21 NO NO19986017A patent/NO312102B1/en not_active IP Right Cessation
-
1999
- 1999-08-16 HK HK99103546A patent/HK1018458A1/en not_active IP Right Cessation
-
2000
- 2000-07-25 US US09/624,966 patent/US6486187B1/en not_active Expired - Lifetime
-
2002
- 2002-09-10 US US10/238,686 patent/US6833375B2/en not_active Expired - Lifetime
-
2004
- 2004-11-02 US US10/979,362 patent/US7205312B2/en not_active Expired - Fee Related
-
2006
- 2006-12-13 US US11/639,043 patent/US7378433B2/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0260744A2 (en) * | 1986-09-15 | 1988-03-23 | Janssen Pharmaceutica N.V. | (1H-imidazol-1-ylmethyl) substituted benzimidazole derivatives |
| EP0371559A2 (en) * | 1988-11-29 | 1990-06-06 | Janssen Pharmaceutica N.V. | Use of benzimidazoles in the treatment of epithelial disorders |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7179825B2 (en) | 1997-12-11 | 2007-02-20 | Janssen Pharmaceutica N.V. | Retinoic acid mimetic anilides |
| US7579352B2 (en) | 1997-12-11 | 2009-08-25 | Janssen Pharmaceutica N.V. | Retinoic acid mimetic anilides |
| US6319939B1 (en) | 1997-12-11 | 2001-11-20 | Janssen Pharmaceutica N.V. | Retinoic acid mimetic anlides |
| WO1999029674A1 (en) * | 1997-12-11 | 1999-06-17 | Janssen Pharmaceutica N.V. | Retinoic acid mimetic anilides |
| US6936626B2 (en) | 1997-12-11 | 2005-08-30 | Janssen Pharmaceutica N.V. | Retinoic acid mimetic anilides |
| US6833373B1 (en) | 1998-12-23 | 2004-12-21 | G.D. Searle & Co. | Method of using an integrin antagonist and one or more antineoplastic agents as a combination therapy in the treatment of neoplasia |
| US6291502B1 (en) | 1999-10-27 | 2001-09-18 | Aventis Pharma Deutschland Gmbh | Use of 2-imidazolyl-substituted carbinols for the production of a medicament for the treatment or phophylaxis of diseases caused by ischemic conditions |
| US7399764B2 (en) | 2002-10-30 | 2008-07-15 | Merck & Co., Inc. | Inhibitors of Akt activity |
| US7638530B2 (en) | 2003-04-24 | 2009-12-29 | Merck & Co., Inc. | Inhibitors of Akt activity |
| US7989462B2 (en) | 2003-07-03 | 2011-08-02 | Myrexis, Inc. | 4-arylamin-or-4-heteroarylamino-quinazolines and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| US8309562B2 (en) | 2003-07-03 | 2012-11-13 | Myrexis, Inc. | Compounds and therapeutical use thereof |
| US8258145B2 (en) | 2005-01-03 | 2012-09-04 | Myrexis, Inc. | Method of treating brain cancer |
| WO2007041379A1 (en) * | 2005-09-30 | 2007-04-12 | Bristol-Myers Squibb Company | Met kinase inhibitors |
| US7547782B2 (en) | 2005-09-30 | 2009-06-16 | Bristol-Myers Squibb Company | Met kinase inhibitors |
| US8513291B2 (en) | 2010-06-01 | 2013-08-20 | Angion Biomedica Corp. | Cytochrome P450 inhibitors and uses thereof |
| US10604533B2 (en) | 2010-11-19 | 2020-03-31 | Ligand Pharmaceuticals Incorporated | Heterocycle amines and uses thereof |
| US11773110B2 (en) | 2010-11-19 | 2023-10-03 | Ligand Pharmaceuticals Incorporated | Heterocycle amines and uses thereof |
| US11186593B2 (en) | 2010-11-19 | 2021-11-30 | Ligand Pharmaceuticals Incorporated | Heterocycle amines and uses thereof |
| US9988374B2 (en) | 2014-08-11 | 2018-06-05 | Angion Biomedica Corp. | Cytochrome P450 inhibitors and uses thereof |
| US10556893B2 (en) | 2014-08-11 | 2020-02-11 | Angion Biomedica Corp. | Cytochrome P450 inhibitors and uses thereof |
| US11459319B2 (en) | 2014-08-11 | 2022-10-04 | Angion Biomedica Corp. | Cytochrome P450 inhibitors and uses thereof |
| US11434234B2 (en) | 2014-12-31 | 2022-09-06 | Angion Biomedica Corp. | Methods and agents for treating disease |
| US10807983B2 (en) | 2015-03-16 | 2020-10-20 | Ligand Pharmaceuticals, Inc. | Imidazo-fused heterocycles and uses thereof |
| US11858938B2 (en) | 2015-03-16 | 2024-01-02 | Ligand Pharmaceuticals, Inc. | Imidazo-fused heterocycles and uses thereof |
| US10414760B2 (en) | 2016-11-29 | 2019-09-17 | Angion Biomedica Corp. | Cytochrome P450 inhibitors and uses thereof |
| CN106588779A (en) * | 2016-12-15 | 2017-04-26 | 青岛辰达生物科技有限公司 | Method for synthesizing dexmedetomidine hydrochloride intermediate |
| CN106632051A (en) * | 2016-12-15 | 2017-05-10 | 青岛辰达生物科技有限公司 | Synthetic method of dexmedetomidine hydrochloride intermediate |
| CN106588780A (en) * | 2016-12-15 | 2017-04-26 | 青岛辰达生物科技有限公司 | Process for preparing dexmedetomidine hydrochloride intermediate |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6833375B2 (en) | N-[4-(heteroarylmethyl)phenyl]-heteroarylamines | |
| US7579352B2 (en) | Retinoic acid mimetic anilides | |
| DE69629300T2 (en) | TRIAZOLIC ANTIFUNGAL AGENTS | |
| AU698199B2 (en) | 6-{triazolyl{3-(trifluoromethyl)phenyl}methyl}-2- quinolinones and -quinolinethiones | |
| US6197972B1 (en) | Triazolones as apolipoprotein-B synthesis inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 97195865.3 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AU BA BB BG BR BY CA CN CU CZ EE GE HU IL IS JP KG KR LC LK LR LT LV MD MG MK MN MX NO NZ PL RO RU SG SI SK TR TT UA US UZ VN AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH KE LS MW SD SZ UG ZW AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1997930378 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1019980709764 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2258165 Country of ref document: CA Ref document number: 2258165 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 333382 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1998-4231 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 178198 Country of ref document: SK |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998/02709 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/1999/000083 Country of ref document: MX |
|
| WWP | Wipo information: published in national office |
Ref document number: 1997930378 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09214080 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1998-4231 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019980709764 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1019980709764 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1997930378 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: PV1998-4231 Country of ref document: CZ |