US20090253842A1 - Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments - Google Patents
Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments Download PDFInfo
- Publication number
- US20090253842A1 US20090253842A1 US12/403,410 US40341009A US2009253842A1 US 20090253842 A1 US20090253842 A1 US 20090253842A1 US 40341009 A US40341009 A US 40341009A US 2009253842 A1 US2009253842 A1 US 2009253842A1
- Authority
- US
- United States
- Prior art keywords
- diol
- diisocyanate
- polyol
- ethyl
- thermoplastic polyurethane
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 0 *[Si]12O[Si]3(*)O[Si]4(*)O[Si](*)(O1)O[Si]1(*)O[Si](*)(O2)O[Si](*)(O3)O[Si](CCC2CCC(O)C(O)C2)(O4)O1.C.C.C.C.C.O=C=NC1=CC=C(CC2=CC=C(N=C=O)C=C2)C=C1.[H]OCCO Chemical compound *[Si]12O[Si]3(*)O[Si]4(*)O[Si](*)(O1)O[Si]1(*)O[Si](*)(O2)O[Si](*)(O3)O[Si](CCC2CCC(O)C(O)C2)(O4)O1.C.C.C.C.C.O=C=NC1=CC=C(CC2=CC=C(N=C=O)C=C2)C=C1.[H]OCCO 0.000 description 6
- HBAZDDTXGRLVMQ-UHFFFAOYSA-N C.C.C.C.C.C.C.C.C.C.C.C.C.C.CC1OC(=O)C(C)OC1=O.O=C1CCCCCO1.[HH].[HH].[H]OCC(=O)OC(C)C(=O)OC(C)C(=O)OCOC(=O)C(C)OC(=O)C(C)OC(=O)CO[H] Chemical compound C.C.C.C.C.C.C.C.C.C.C.C.C.C.CC1OC(=O)C(C)OC1=O.O=C1CCCCCO1.[HH].[HH].[H]OCC(=O)OC(C)C(=O)OC(C)C(=O)OCOC(=O)C(C)OC(=O)C(C)OC(=O)CO[H] HBAZDDTXGRLVMQ-UHFFFAOYSA-N 0.000 description 1
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/30—Low-molecular-weight compounds
- C08G18/38—Low-molecular-weight compounds having heteroatoms other than oxygen
- C08G18/3893—Low-molecular-weight compounds having heteroatoms other than oxygen containing silicon
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/42—Polycondensates having carboxylic or carbonic ester groups in the main chain
- C08G18/4266—Polycondensates having carboxylic or carbonic ester groups in the main chain prepared from hydroxycarboxylic acids and/or lactones
- C08G18/4269—Lactones
- C08G18/4277—Caprolactone and/or substituted caprolactone
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/42—Polycondensates having carboxylic or carbonic ester groups in the main chain
- C08G18/4266—Polycondensates having carboxylic or carbonic ester groups in the main chain prepared from hydroxycarboxylic acids and/or lactones
- C08G18/428—Lactides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/48—Polyethers
- C08G18/4833—Polyethers containing oxyethylene units
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/61—Polysiloxanes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/02—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes
- C08G61/04—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes only aliphatic carbon atoms
- C08G61/06—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes only aliphatic carbon atoms prepared by ring-opening of carbocyclic compounds
- C08G61/08—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes only aliphatic carbon atoms prepared by ring-opening of carbocyclic compounds of carbocyclic compounds containing one or more carbon-to-carbon double bonds in the ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L65/00—Compositions of macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain; Compositions of derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2230/00—Compositions for preparing biodegradable polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2280/00—Compositions for creating shape memory
Definitions
- the instant disclosure relates to shape memory polymers and more particularly thermoplastic polyurethanes with an alternating sequence of hard and soft segments in which a nanostructured polyhedral oligosilsesquioxane diol is used as a chain extender to form a crystalline hard segment and also relates to methods for the preparation of these thermoplastic polyurethanes and to applications thereof.
- Shape memory materials feature an ability to transform shape from a temporary, frozen, shape to a permanent shape when triggered by an environmental stimulus, such as heat, light, or vapor. Used creatively, these phenomena can be exploited for a wide range of applications. While both shape memory alloys (SMAs) and shape memory polymers (SMPs) show similar thermo-stimulated shape memory properties, their mechanisms of action are quite distinct. Advantages of SMAs include rapid strain recovery (within 1 second), the potential training for two-way reversible memory, and an apparent superelasticity due within the austentite phase at low temperature.
- polymers intrinsically exhibit shape memory effects derived from their highly coiled constituent chains that are collectively extensible via mechanical work and this energy may be stored indefinitely, known as “shape fixing,” by cooling below T g or T m .
- shape fixing by cooling below T g or T m .
- the polymeric samples can later perform mechanical work and return to a stress-free state when heated above the critical temperature, mobilizing the frozen chains to regain the entropy of their coiled state.
- thermally stimulated SMPs have the advantages of: (i) large recoverable deformations in excess of several hundred percent strain; (ii) facile tuning of transition temperatures through variation of the polymer chemistry; and (iii) processing ease at low cost.
- Thermally stimulated SMPs with different thermomechanical properties to function in various applications have previously been synthesized and characterized by the instant inventors.
- the materials span a range of room temperature moduli, from rigid glassy materials having storage moduli of several GPa to compliant rubbers with moduli as low as tens of MPa.
- the retracting (rubbery) moduli have been adjusted over the range 0.5 ⁇ E ⁇ 10 MPa, as prescribed by the end application.
- One such example is chemically crosslinked polycyclooctene (PCO), a stiff semicrystalline rubber that is elastically deformed above T m to a temporary shape that is fixed by crystallization. Fast and complete recovery of gross deformations is achieved by immersion in hot water.
- PCO polycyclooctene
- miscible blends of a semicrystalline polymer with amorphous polymers have also been intensively investigated due to their attractive crystalline properties and mechanical properties. For those blends that are miscible at the molecular level, a single glass transition results, without broadening, an aspect important to shape memory. Additionally, in such miscible blends the equilibrium crystallinity (which controls the plateau modulus between T g and T m where shape fixing is performed) also changes dramatically and systematically with the blend compositions. It provides a simple route to alternative shape memory plastics; i.e.
- Microphase-separated semicrystalline thermoplastic polymers with two sharp melting transitions T m2 >T m1 >room temperature, where the difference of the two melting points is at least 20° C. are also good candidates for shape memory offering the advantage of melt processing above T m2 , and repeated resetting of the equilibrium shape by relaxing stress in the fluid state.
- Representative past examples of such polymers in this class of SMP are conventional polyurethanes whose soft domains are glassy or semicrystalline with low melting point (but higher than T crit ) and whose hard domains feature a higher melting point only exceeded during processing.
- a further object of the invention is to provide hybrid polyurethanes that are biocompatible and can be used as medical devices and implants.
- Still a further object of the invention is to provide hybrid polyurethanes that are biodegradable and whose biodegradation rate can be controlled by the type of polyol, the molecular weight of polyol, and the POSS content.
- Still a further object of the invention is to provide hybrid polyurethanes that can be used as a drug delivery vehicle whose elution profile is controlled by the polymer composition; specifically, the chemical structures, the molecular weight, and the weight ratio of the polyol in the polyurethane.
- Yet another object of the invention is a method for synthesizing such hybrid polyurethanes.
- the disclosure provides a method for producing hybrid polyurethane SMPs by reacting (A) a polyol, (B) a chain extender dihydroxyl-terminated polyhedral oligosilsesquioxane (dihydroxyl-terminated POSS) and (C) a diisocyanate.
- a polyol is defined as a polymeric diol.
- the polyol (A) can be a nonbiodegradable one, such as, for example: polyethylene glycol (PEG), polytetrahydrofuran (polyTHF), and diols prepared from polycyclooctene (PCO), trans-1,4 butadiene, or trans-isoprene; or a biodegradable one, such as, for example: diols prepared from caprolactone (polycaprolactone (PCL) diol), polycaprolactone-polylactide random copolymers, polycaprolactone-polyglycolide random copolymers, polycaprolactone-polylactide-polyglycolide random copolymers, polylactide polyol, polycaprolactone-poly( ⁇ -hydroxybutyric acid) random copolymers, or poly( ⁇ -hydroxybutyric acid).
- PEG polyethylene glycol
- polyTHF polytetrahydrofuran
- PCO polycycloocten
- the diols prepared from caprolactone include, for example, diols obtained from the polymerization of caprolactone initiated with a low molecular weight diol to obtain a polycaprolactone (PCL) diol.
- Suitable low molecular weight diol initiators include, for example, C 1 -C 10 alkyl diols (e.g. propane diol, butane diol, etc.).
- Hydroxyl-terminated poly(trans-1,4-butadiene), hydroxyl-terminated polycyclooctene (PCO diol), and hydroxyl-terminated poly(trans-1,4-isoprene) can also be prepared by methods known by one of ordinary skill in the art. See, e.g.
- hydroxyl-terminated polymethacrylate copolymers for example diols of polymethyl methacrylate (PMMA) copolymerized with a T g -reducing comonomer, including methyl, ethyl, propyl, or butyl (meth)acrylate.
- PMMA polymethyl methacrylate
- T g -reducing comonomer including methyl, ethyl, propyl, or butyl (meth)acrylate.
- the hydroxyl-terminated poly(methyl)methacrylate copolymers can be prepared via controlled radical polymerization methods. An example of the synthesis of hydroxyl-terminated poly(meth)acrylate copolymers can be found in Macromolecules 37 (2004) pp. 9694-9700.
- the chain extender dihydroxyl-terminated POSS (B) can be a compound containing a polyhedral oligosilsesquioxane moiety and a diol moiety, wherein a linking group links the two moieties.
- Commercially available polyhedral oligosilsesquioxane diols include those provided by Hybrid PlasticsTM Hattiesburg, Miss. or Aldrich Chemical (see generally “ Silsesquioxanes, Bridging the Gap Between Polymers and Ceramics ”, Chemfiles, Vol. 1, No. 6, 2001 (Aldrich Chemical).
- Exemplary polyhedral oligosilsesquioxane diols include 1-(2,3-propanediol)propoxy-3,5,7,9,11,13,15-isobutylpentacyclo-[9.5.1.1 3,9 .1 5,15 .1 7,13 ]octasiloxane (“1,2-propanediolisobutyl-POSS” CAS # 480-439-49-4); 1-(2,3-propanediol)propoxy-3,5,7,9,11,13,15-cyclohexylpentacyclo-[9.5.1.1 3,9 .1 5,15 .1 7,13 ]octasiloxane (“1,2-propanediolcyclohexyl-POSS”); 2-ethyl-2-[3-[[(heptacyclopentylpentacyclo-[9.5.1.1 3,9 .1 5,15 .1 7,13 ]octa
- the diisocyanate (C) can be selected from a large number of diisocyanates and is preferably 4,4′-diphenyl methylene diisocyanate (MDI).
- MDI 4,4′-diphenyl methylene diisocyanate
- Other diisocyanates (C) that will function well for the synthesis of hybrid polyurethane SMPs include, for example: toluene-2,4-diisocyanate (TDI), toluene-2,6-diisocyanate, hexamethylene-1,6-diisocyanate (HDI), isophorone diisocyanate (IPDI), hydrogenated 4,4′-diphenylmethane diisocyanate (H12MDI), 1,3-bis-isocyanato-1-methylene ethylene benzene, and the like.
- TDI toluene-2,4-diisocyanate
- HDI hexamethylene-1,6-diiso
- the (B) list can be used in conjunction with non-hybrid (conventional) low molecular weight diols. These can be chosen from alkane diols (e.g., 1,3-propanediol, 1,4-butane diol, 1,5-n-pentane diol, 1,6-n-hexane diol, 1,4-trans cyclohexane exodiol, and 1,4-trans cyclohexane endodiol).
- alkane diols e.g., 1,3-propanediol, 1,4-butane diol, 1,5-n-pentane diol, 1,6-n-hexane diol, 1,4-trans cyclohexane exodiol, and 1,4-trans cyclohexane endodiol.
- the polyol can be semicrystalline and preferably selected from polyethylene glycol (PEG), hydroxyl-terminated polycaprolactone (PCL), hydroxyl-terminated polycyclooctene (PCO), hydroxyl-terminated poly(trans-1,4-butadiene), hydroxyl-terminated poly(trans-isoprene) or it can be amorphous in which case it can be poly(tetrahydrofuran) diol, polynorbornene diol and/or a hydroxyl-terminated poly(methyl)methacrylate copolymer or homopolymer.
- PEG polyethylene glycol
- PCL hydroxyl-terminated polycaprolactone
- PCO hydroxyl-terminated polycyclooctene
- trans-1,4-butadiene hydroxyl-terminated poly(trans-isoprene)
- it can be amorphous in which case it can be poly(tetrahydrofuran) diol, polynorbornen
- microcrystalline is defined as the physical state where at least a portion of the material is spatially organized into crystalline regions that are characterized by both a distinct crystalline structure and a melting transition, T m , above which the material behaves as a structure-less liquid.
- diisocyanates (C) include, for example, C 4 -C 30 linear or branched alkyl diisocyanates; C 8 -C 30 aryl diisocyanates including diisocyanates containing phenyl groups; and the like.
- the alkyl or aryl groups can be substituted with one or more substituents chosen from C 4 -C 10 tertiary alkyl, C 1 -C 12 primary or secondary alkyl, C 4 -C 10 tertiary alkoxy, C 1 -C 12 primary or secondary alkoxy, halogen, and the like.
- the mol ratio of polyol:chain extender:diisocyanate can be about 1:2:3; specifically about 1:5:6; and more specifically about 1:10:11.
- TPUs can be prepared from a ratio of polyol to polyhedral oligosilsesquioxane diol (ratio of x:y) of about 1:2 to about 1:20, specifically about 1:4 to about 1:12, and more specifically about 1:5 to about 1:10.
- the ratio of polyol to dihydroxyl-terminated POSS affects the shape memory properties of the resulting TPU by determining the flatness of the rubber modulus versus temperature plateau above the T m or T g of the polyol segment.
- the shape memory polymer comprises a T g above about 37° C., specifically above about 55° C.
- Scheme 1 shows an example of the synthesis of TPU using polyethylene glycol as polyol (n is a number such that the diol molecular weight is in the range of about 2,000 to about 20,000 g/mol), trans-cyclohexanediol isobutyl-POSS as chain extender to react with 4,4′ diphenyl methylene diisocyanate in toluene.
- Scheme 2 shows an example of the synthesis of TPU using a polycaprolactone diol as polyol (n is a number such that 2*n gives a total PCL diol molecular weight in the range of about 2,000 to about 20,000 g/mol), TMP Isobutyldiol-POSS as chain extender to react with 4,4′ diphenyl methylene diisocyanate.
- Scheme 3 shows an example of the synthesis of TPU using a polycyclooctene diol as polyol (n is chosen such that the diol molecular weight is in the range of about 2,000 to about 20,000 g/mol), TMP Isobutyldiol-POSS as chain extender to react with 4,4′ diphenyl methylene diisocyanate.
- Scheme 4 illustrates an example of the synthesis of polycaprolactone-polylactide random copolymer diol using ring-opening polymerization.
- the copolymers show one single sharp T g that can be tunable according to the CL:LA molar ratios.
- D,L-lactide or “meso” lactide (CAS # 96-95-5,3,6-dimethyl-1,4-dioxane-2,5-dione) is reacted with caprolactone to provide the copolymer.
- Scheme 5 shows an example of synthesis of TPU using a polycaprolactone-D,L-polylactide random copolymer as polyol, TMP Isobutyldiol-POSS(R is isobutyl) as chain extender to react with 4,4′ diphenyl methylene diisocyanate.
- the R substituent can include a C 1 -C 12 primary, secondary, or tertiary alkyl group.
- exemplary R groups include methyl, isobutyl, isooctyl, cyclopentyl, cyclohexyl, phenyl, and the like.
- the instant hybrid polyurethanes demonstrate sharp and tunable transition temperatures, adjustable stiffness above their transition temperatures, and thermal processability above the melting point of the POSS domains.
- the hybrid polyurethanes also show excellent shape recovery effect at the recovery temperature and the retracting force is adjustable according to the composition of the POSS. They also posses a unique property that is different from the other shape memory polymers in that the current invention (in the PEG embodiment) can be triggered to recover by moisture (liquid or vapor) aside from heating.
- the range 30° C. to 60° C. according to the ratio of the components used and (importantly) thermal annealing to achieve steady-state (equilibrium) crystallinity is important. The recovery can be finished within seconds when heated 20° C.
- the materials are rigid at room temperature, the polymers generally are biocompatible.
- the described TPUs are biodegradable, and the biodegradation rate can be controlled by the chemical compositions, soft segment length, and soft-segment/hard-segment ratios, and can be used as medical devices and implants, as the degradation products are generally non-toxic, non-immunogenic, and absorbable.
- Biodegradability can be estimated by measuring the percent mass loss after 70 days immersion in a buffered saline solution (0.01 M phosphate, 0.138 M NaCl, 0.027 M KCl). This test is described in detail in the working examples below.
- the materials can also be used as drug elution stents or stent coatings.
- the materials can further be dyed to any color or rendered radio-opaque for x-ray radiography according to application requirements.
- any of the hybrid polyurethane polymers mentioned above may be filled with, for example, nanoparticles of boron nitride, silica, titanium dioxide, montmorillonite, clay, Kevlar, staple, aluminum nitride, barium subcarbonate and bismuth subcarbonate. Clay and silica can be used to, for example, increase the modulus of the plastic.
- Dispersing agents and/or compatibilizing agents may be used, for example, to improve the blending of polymers and the blending of polymers with fillers.
- Dispersing agents and/or compatibilizing agents include, for example, ACRAWAX® (ethylene bis-stearamide), polyurethanes and ELVALOY® (acrylic functionalized polyethylene).
- the rate of biodegradation of the thermoplastic polyurethane shape memory polymer can be controlled by adjusting the content of POSS chain extender, the content and composition of the polyol.
- the biodegradation rate can be a) decreased by increasing the amount of POSS chain extender in the polymer; b) decreased by increasing the molecular weight of the polyol in the polymer; or c) increased by increasing the amount of hydrolysable groups in the polyol.
- a drug eluting implant, a drug eluting stent, or drug eluting stent coating is prepared from a biodegradable shape memory polymer.
- a thermoplastic polyurethane shape memory polymer can exhibit a certain rate of biodegradation. The rate of drug elution from the polymer correlates to the rate of biodegradation of the shape memory polymer.
- FIG. 1 illustrates graphically the DMA plots of the TMP POSS based thermoplastic polyurethane (TPU) with mole ratio of PEG:POSS as 1:6, 1:4 respectively;
- FIG. 2 illustrates graphically the DSC results of TMP POSS based TPU with different PEG:POSS mole ratios
- FIG. 5 illustrates graphically the mass loss of TPUs with different soft segment length and soft-segment/hard-segment ratios in phosphate buffered saline;
- FIG. 6 illustrates graphically the molecular weight decrease of TPUs with different soft segment length and soft-segment/hard-segment ratios in phosphate buffered saline
- FIG. 7 illustrates graphically the critical temperature change of a TPU in phosphate buffered saline.
- Thermoplastic polyurethanes with different compositions were synthesized by one-step condensation polymerization using scheme 1 shown above ((A) PEG as the polyol, (B) transcyclohexanediolisobutyl-polyhedral oligosilsesquioxane diol and (C) MDI as the diisocyanate.
- Toluene was used as solvent and dibutyltin dilaurate was used as catalyst.
- the reaction was kept at 90° C. under the nitrogen for 2 hours and then cooled down to room temperature and precipitated into hexane.
- the product was dried thoroughly and dissolved in toluene to make a 10 wt % solution for casting films.
- Table 1 The molecular weights and molecular weight distributions of this series of samples obtained from size exclusion chromatography are summarized in Table 1.
- the melting temperature of the soft segment is observed to shift to lower values with a broadening of the melting peak while the melting temperature of the hard segment is observed to shift to higher values with a sharpening of the melting peak when the mole ratio of polyol:chain extender decreases.
- This result can be explained in that as the PEG:POSS ratio decreases, the resulting block copolymer will have less overall PEG content, which will directly affect the size and perfection of the crystallization of PEG blocks. Therefore, the melting temperature moves to lower values and the peak is broadened.
- the content of POSS will increase in the block copolymers, which provides for more clear aggregation of hard segments to form larger and more perfect crystals. Therefore, the melting temperature of hard segment moves to higher values while the peak is sharpened ( FIG. 2 ).
- the dried films of the formed polyurethanes were cut into thin strips for tests of temporary shape fixing and subsequent recovery, or shape memory. For example, a sample was first heated on the hot stage to 65° C., which is well above the first transition temperature but low enough to avoid melting of the elastic network of the POSS-rich phase. It was then stretched to a certain degree of elongation and cooled down to the room temperature. The deformed shape was fixed at room temperature. Finally, the deformed sample was heated up again on hot plate to 65° C. and it was observed that the sample restored to its original length completely and within seconds. A similar phenomenon was observed when water was used as a stimulus for the shape recovery except that the sample secondarily swelled to form a tough hydrogel.
- Biodegradation studies were performed on polyurethanes prepared from poly( ⁇ -caprolactone)-co-poly(D,L-lactide) as the polyol; 1-[2-ethyl-2-[(3-dimethylsiloxy)propoxymethyl]-1,3-propanediol]-3,5,7,9,11,13,15-isobutylpentacyclo-[9.5.1 13,9 .1 5,15 .1 7,13 ]-octasiloxane as the dihydroxyl-terminated POSS; and 4,4-methylenebis(phenyl-isocyanate) as the diisocyanate according to the following procedure.
- PCL-co-PDLLA poly( ⁇ -caprolactone)-co-poly(D,L-lactide)
- PCL-co-PDLLA poly( ⁇ -caprolactone)-co-poly(D,L-lactide)
- a mixture of ⁇ -caprolactone (99%, Aldrich, used as received), D, L-lactide (99%, Aldrich, purified by recrystallization), 1,4-butane diol (96%, Aldrich, dried using 4 ⁇ molecular sieves) (molar amounts varied) and catalytic stannous octoate were put in a three-neck flask equipped with nitrogen inlet and outlet. Magnetic stirring was used to agitate the reaction mixture. The temperature was set at 140° C. and the polymerization reaction took 4 hours to complete. The product was dissolved in toluene and precipitated into hexane. The sample of PCL-co-PDLLA was dried in vacuum oven overnight.
- thermoplastic polyurethanes were then synthesized using a one-step polymerization method.
- a 50 mL three-neck flask equipped with nitrogen inlet and outlet, condenser and thermometer was used as the reactor for the polymerization.
- the reactor was heated to 50° C., followed by adding 0.2633 g (1.05 ⁇ 10 ⁇ 3 mol) of 4,4-methylenebis(phenyl-isocyanate) (98%, Aldrich, used as received).
- the reaction mixture was then heated to 90° C. and 2 drops of dibutyltin dilaurate (95%, Aldrich) was added through a syringe.
- a thickening phenomenon was observed during the reaction and the reaction was kept at 90° C. for 2 hr under the nitrogen atmosphere before completion. Then the thickened polymer solution was precipitated into excess amount of n-hexane, filtered and dried to remove any unreacted POSS.
- the dried polymer was dissolved in toluene again to make a 10 wt % of solution for casting films.
- Several series of samples were made using PCL-co-PDLLA as soft segment and the ratio of PCL-co-PDLLA/POSS as variables (See FIG. 5 ).
- Biodegradation test of samples (approximately 150 micrometers thick) with different polyol molecular weight and different polyol:POSS ratio was carried out at 37° C. in Phosphate Buffered Saline (PBS) ((0.01 M); NaCl 0.138 M; KCl 0.0027 M) buffer.
- PBS Phosphate Buffered Saline
- the samples were put in the buffer for 92 days and the progress of degradation was measured every 10 days by taking the samples from the buffer and determining the percent mass retained.
- the data are summarized in FIG. 5 .
- the degradation was very slow at the first 30 days.
- the samples having higher molecular weight of soft segment degraded more slowly than the ones having low molecular weight of soft segment.
- those having higher POSS content degraded more slowly than the ones having lower POSS content. Therefore, it can be speculated that the soft segment is a dominant species at the beginning of the degradation.
- the degradation progressed by chain scission of the soft segment, particularly hydrolysis of the ester linkages (Schemes 4 and 5 ). Therefore, the longer the soft segment, the slower the rate of degradation.
- the weight fraction of the POSS is higher, the same chain scission is suppressed and the rate of degradation will also be decreased.
- SEC Size Exclusion Chromatography
- the hybrid polyurethanes of the invention can be used for the following applications.
- shape memory polymers of the disclosure are particularly suitable as biomaterials because of their low thrombogenicity, high biocompatibility, as well as unique mechanical properties.
- shape memory polyurethanes were formulated such that the melting temperature of one segment falls within a useful temperature range for biomedical application: 37° C.-50° C.
- the present disclosure provides an advantageous shape memory polymer that includes thermoplastic polyurethane shape memory polymers formed by reacting in one step a polyol, a POSS chain extender and a diisocyanate, having medium and tunable modulus in the fixed state at room temperature having a tunable sharp transition, whose permanent shape can be repeatedly remolded above a certain melting temperature.
Abstract
Thermoplastic polyurethanes having an alternating sequence of hard and soft segments in which a nanostructured polyhedral oligosilsesquioxane diol is used as a chain extender to form a crystalline hard segment constituting SMPs. The polyurethanes are formed by reacting a polyol, a chain extender dihydroxyl-terminated polyhedral oligosilsesquioxane and a diisocyanate. The polyurethanes have multiple applications including for example, implants for human health care, drug delivery matrices, superabsorbant hydrogels, coatings, adhesives, temperature and moisture sensors, etc.
Description
- This application is a continuation of U.S. Nonprovisional patent application Ser. No. 11/111,388 filed Apr. 21, 2005, which is a continuation-in-part application of U.S. Nonprovisional patent application Ser. No. 10/683,167 filed Oct. 10, 2003 and issued Aug. 15, 2006 as U.S. Pat. No. 7,091,297 B2, which claims benefit of U.S. Provisional Application Nos. 60/418,023 filed Oct. 11, 2002, 60/466,401 filed Apr. 29, 2003, 60/419,506 filed Oct. 18, 2002, 60/488,590 filed Jul. 18, 2003, and 60/488,323 filed Jul. 18, 2003; each of which is incorporated herein by reference in its entirety. U.S. Nonprovisional patent application Ser. No. 10/425,451 filed Apr. 29, 2003, and U.S. Provisional Patent Application No. 60/377,544 filed May 2, 2008, to which it claims priority, are also incorporated herein by reference.
- The instant disclosure relates to shape memory polymers and more particularly thermoplastic polyurethanes with an alternating sequence of hard and soft segments in which a nanostructured polyhedral oligosilsesquioxane diol is used as a chain extender to form a crystalline hard segment and also relates to methods for the preparation of these thermoplastic polyurethanes and to applications thereof.
- Shape memory materials feature an ability to transform shape from a temporary, frozen, shape to a permanent shape when triggered by an environmental stimulus, such as heat, light, or vapor. Used creatively, these phenomena can be exploited for a wide range of applications. While both shape memory alloys (SMAs) and shape memory polymers (SMPs) show similar thermo-stimulated shape memory properties, their mechanisms of action are quite distinct. Advantages of SMAs include rapid strain recovery (within 1 second), the potential training for two-way reversible memory, and an apparent superelasticity due within the austentite phase at low temperature. In contrast, polymers intrinsically exhibit shape memory effects derived from their highly coiled constituent chains that are collectively extensible via mechanical work and this energy may be stored indefinitely, known as “shape fixing,” by cooling below Tg or Tm. The polymeric samples can later perform mechanical work and return to a stress-free state when heated above the critical temperature, mobilizing the frozen chains to regain the entropy of their coiled state. In comparison to SMAs, thermally stimulated SMPs have the advantages of: (i) large recoverable deformations in excess of several hundred percent strain; (ii) facile tuning of transition temperatures through variation of the polymer chemistry; and (iii) processing ease at low cost.
- Thermally stimulated SMPs with different thermomechanical properties to function in various applications, for example as medical devices and mechanical actuators have previously been synthesized and characterized by the instant inventors. The materials span a range of room temperature moduli, from rigid glassy materials having storage moduli of several GPa to compliant rubbers with moduli as low as tens of MPa. Moreover, the retracting (rubbery) moduli have been adjusted over the range 0.5<E<10 MPa, as prescribed by the end application. One such example is chemically crosslinked polycyclooctene (PCO), a stiff semicrystalline rubber that is elastically deformed above Tm to a temporary shape that is fixed by crystallization. Fast and complete recovery of gross deformations is achieved by immersion in hot water. These SMPs have been described in Provisional Patent Application Ser. No. 60/419,506 filed Oct. 18, 2002 entitled Chemically Crosslinked Polycyclooctene, the entirety of which is incorporated herein by reference. In Provisional Patent Application Ser. No. 60/377,544 filed May 2, 2002 entitled Castable Shape Memory Polymers, the entirety of which is incorporated herein by reference, stiffer SMPs offering tunable critical temperatures and rubber modulus using a thermosetting random copolymer made of two vinyl monomers that yield controlled Tg and casting-type processing are described. Such copolymers were crosslinked with a difunctional vinyl monomer (crosslinker), the concentration of crosslinker controlling the rubber modulus and thus the work potential during recovery. Besides their shape memory effects, these materials are also castable allowing for processing more complex shapes. In addition, they are optically transparent making them useful for additional applications.
- The use of chemical crosslinking in both of these cases limits the types of processing possible and forever sets the equilibrium shape at the point of network formation. Therefore, miscible blends of a semicrystalline polymer with amorphous polymers have also been intensively investigated due to their attractive crystalline properties and mechanical properties. For those blends that are miscible at the molecular level, a single glass transition results, without broadening, an aspect important to shape memory. Additionally, in such miscible blends the equilibrium crystallinity (which controls the plateau modulus between Tg and Tm where shape fixing is performed) also changes dramatically and systematically with the blend compositions. It provides a simple route to alternative shape memory plastics; i.e. SMPs with relatively high modulus in the fixed state at room temperature, having a tunable and sharp transition, and the permanent shape can be remolded repeatedly above certain melting temperatures. These SMP blends have been described in Provisional Patent Application Ser. No. 60/466,401 filed Apr. 29, 2003 entitled Blends of Amorphous and Semicrystalline Polymers with Shape Memory Properties, the entirety of which is incorporated herein by reference.
- Microphase-separated semicrystalline thermoplastic polymers with two sharp melting transitions Tm2>Tm1>room temperature, where the difference of the two melting points is at least 20° C., are also good candidates for shape memory offering the advantage of melt processing above Tm2, and repeated resetting of the equilibrium shape by relaxing stress in the fluid state. Representative past examples of such polymers in this class of SMP are conventional polyurethanes whose soft domains are glassy or semicrystalline with low melting point (but higher than Tcrit) and whose hard domains feature a higher melting point only exceeded during processing.
- It is an object of the present invention to provide shape memory polymers comprising hybrid polyurethanes.
- It is another object of the invention to provide shape memory polymers having medium and tunable modulus in the fixed state at room temperature, having a tunable and sharp transition, whose permanent shape can be repeatedly remolded above a certain melting temperature.
- It is another object of the invention to provide hybrid polyurethane SMPs evidencing sharp and tunable transition temperatures, adjustable stiffness above their transition temperatures, especially physical properties at 37° C. controlled by the level of POSS content, and thermal processability above the melting point of the POSS domains.
- It is yet another object of the invention to provide hybrid polyurethane SMPs which possess excellent shape recovery effect at the recovery temperature and the retracting force is adjustable according to the composition of the POSS.
- A further object of the invention is to provide hybrid polyurethanes that are biocompatible and can be used as medical devices and implants.
- Still a further object of the invention is to provide hybrid polyurethanes that are biodegradable and whose biodegradation rate can be controlled by the type of polyol, the molecular weight of polyol, and the POSS content.
- Still a further object of the invention is to provide hybrid polyurethanes that can be used as a drug delivery vehicle whose elution profile is controlled by the polymer composition; specifically, the chemical structures, the molecular weight, and the weight ratio of the polyol in the polyurethane.
- Yet another object of the invention is a method for synthesizing such hybrid polyurethanes.
- Broadly the disclosure provides a method for producing hybrid polyurethane SMPs by reacting (A) a polyol, (B) a chain extender dihydroxyl-terminated polyhedral oligosilsesquioxane (dihydroxyl-terminated POSS) and (C) a diisocyanate. As used herein, the term “polyol” is defined as a polymeric diol. The polyol (A) can be a nonbiodegradable one, such as, for example: polyethylene glycol (PEG), polytetrahydrofuran (polyTHF), and diols prepared from polycyclooctene (PCO), trans-1,4 butadiene, or trans-isoprene; or a biodegradable one, such as, for example: diols prepared from caprolactone (polycaprolactone (PCL) diol), polycaprolactone-polylactide random copolymers, polycaprolactone-polyglycolide random copolymers, polycaprolactone-polylactide-polyglycolide random copolymers, polylactide polyol, polycaprolactone-poly(β-hydroxybutyric acid) random copolymers, or poly(β-hydroxybutyric acid). The diols prepared from caprolactone include, for example, diols obtained from the polymerization of caprolactone initiated with a low molecular weight diol to obtain a polycaprolactone (PCL) diol. Suitable low molecular weight diol initiators include, for example, C1-C10 alkyl diols (e.g. propane diol, butane diol, etc.). Hydroxyl-terminated poly(trans-1,4-butadiene), hydroxyl-terminated polycyclooctene (PCO diol), and hydroxyl-terminated poly(trans-1,4-isoprene) can also be prepared by methods known by one of ordinary skill in the art. See, e.g. Polymer 42 (2001) pp. 4939-4945, for the preparation of diols via ring opening metathesis-polymerization chemistry. Eur. Polym. J. (1995) 31:51 and Eur. Polym. J. (1997) 31:339) disclose methods to prepare hydroxyl-terminated poly(isoprene). Sartomer Co. Inc. of Exton PA provides several commercially available hydroxyl terminated polybutadienes.
- Also contemplated herein are hydroxyl-terminated polymethacrylate copolymers, for example diols of polymethyl methacrylate (PMMA) copolymerized with a Tg-reducing comonomer, including methyl, ethyl, propyl, or butyl (meth)acrylate. The hydroxyl-terminated poly(methyl)methacrylate copolymers can be prepared via controlled radical polymerization methods. An example of the synthesis of hydroxyl-terminated poly(meth)acrylate copolymers can be found in Macromolecules 37 (2004) pp. 9694-9700.
- The chain extender dihydroxyl-terminated POSS (B) can be a compound containing a polyhedral oligosilsesquioxane moiety and a diol moiety, wherein a linking group links the two moieties. Commercially available polyhedral oligosilsesquioxane diols include those provided by Hybrid Plastics™ Hattiesburg, Miss. or Aldrich Chemical (see generally “Silsesquioxanes, Bridging the Gap Between Polymers and Ceramics”, Chemfiles, Vol. 1, No. 6, 2001 (Aldrich Chemical). Exemplary polyhedral oligosilsesquioxane diols include 1-(2,3-propanediol)propoxy-3,5,7,9,11,13,15-isobutylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxane (“1,2-propanediolisobutyl-POSS” CAS # 480-439-49-4); 1-(2,3-propanediol)propoxy-3,5,7,9,11,13,15-cyclohexylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxane (“1,2-propanediolcyclohexyl-POSS”); 2-ethyl-2-[3-[[(heptacyclopentylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxanyl)oxy]dimethylsilyl]-propoxy]methyl]-1,3-propanediol (“TMP cyclopentyldiol-POSS” or “TMP Diolcyclopentyl-POSS”, CAS 268747-51-9); 2-ethyl-2-[3-[[(heptacyclohexylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxanyl)oxy]dimethylsilyl]-propoxy]methyl]-1,3-propanediol (“TMP cyclohexyldiol-POSS”); 2-ethyl-2-[3-[[(heptaisobutylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxanyl)oxy]dimethylsilyl]-propoxy]methyl]-1,3-propanediol (“TMP isobutyldiol-POSS” or “TMP diolisobutyl-POSS”); 1-(2-trans-cyclohexanediol)ethyl-3,5,7,9,11,13,15-cyclohexanepentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxane (“trans-cyclohexanediolcyclohexane-POSS” or “trans-cyclohexanediolcyclohexyl-POSS”); 1-(2-trans-cyclohexanediol)ethyl-3,5,7,9,11,13,15-isobutylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxane, (“transcyclohexanediolisobutyl-POSS”, CAS 480-439-48-3); and 2-ethyl-2-[3-[[(heptaisobutylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxanyl)oxy]-dimethylsilyl]propoxy]propane-1,3-diol.
- The diisocyanate (C) can be selected from a large number of diisocyanates and is preferably 4,4′-diphenyl methylene diisocyanate (MDI). Other diisocyanates (C) that will function well for the synthesis of hybrid polyurethane SMPs include, for example: toluene-2,4-diisocyanate (TDI), toluene-2,6-diisocyanate, hexamethylene-1,6-diisocyanate (HDI), isophorone diisocyanate (IPDI), hydrogenated 4,4′-diphenylmethane diisocyanate (H12MDI), 1,3-bis-isocyanato-1-methylene ethylene benzene, and the like.
- The (B) list can be used in conjunction with non-hybrid (conventional) low molecular weight diols. These can be chosen from alkane diols (e.g., 1,3-propanediol, 1,4-butane diol, 1,5-n-pentane diol, 1,6-n-hexane diol, 1,4-trans cyclohexane exodiol, and 1,4-trans cyclohexane endodiol).
- The polyol can be semicrystalline and preferably selected from polyethylene glycol (PEG), hydroxyl-terminated polycaprolactone (PCL), hydroxyl-terminated polycyclooctene (PCO), hydroxyl-terminated poly(trans-1,4-butadiene), hydroxyl-terminated poly(trans-isoprene) or it can be amorphous in which case it can be poly(tetrahydrofuran) diol, polynorbornene diol and/or a hydroxyl-terminated poly(methyl)methacrylate copolymer or homopolymer. As used herein, “semicrystalline” is defined as the physical state where at least a portion of the material is spatially organized into crystalline regions that are characterized by both a distinct crystalline structure and a melting transition, Tm, above which the material behaves as a structure-less liquid.
- Other suitable diisocyanates (C) include, for example, C4-C30 linear or branched alkyl diisocyanates; C8-C30 aryl diisocyanates including diisocyanates containing phenyl groups; and the like. Optionally the alkyl or aryl groups can be substituted with one or more substituents chosen from C4-C10 tertiary alkyl, C1-C12 primary or secondary alkyl, C4-C10 tertiary alkoxy, C1-C12 primary or secondary alkoxy, halogen, and the like. The mol ratio of polyol:chain extender:diisocyanate can be about 1:2:3; specifically about 1:5:6; and more specifically about 1:10:11. TPUs can be prepared from a ratio of polyol to polyhedral oligosilsesquioxane diol (ratio of x:y) of about 1:2 to about 1:20, specifically about 1:4 to about 1:12, and more specifically about 1:5 to about 1:10. The ratio of polyol to dihydroxyl-terminated POSS affects the shape memory properties of the resulting TPU by determining the flatness of the rubber modulus versus temperature plateau above the Tm or Tg of the polyol segment. In some embodiments, the shape memory polymer comprises a Tg above about 37° C., specifically above about 55° C.
- The method of the invention and the novel hybrid polyurethanes prepared thereby are illustrated by the following non-limiting reaction schemes.
-

-
Scheme 1 shows an example of the synthesis of TPU using polyethylene glycol as polyol (n is a number such that the diol molecular weight is in the range of about 2,000 to about 20,000 g/mol), trans-cyclohexanediol isobutyl-POSS as chain extender to react with 4,4′ diphenyl methylene diisocyanate in toluene. -

-
Scheme 2 shows an example of the synthesis of TPU using a polycaprolactone diol as polyol (n is a number such that 2*n gives a total PCL diol molecular weight in the range of about 2,000 to about 20,000 g/mol), TMP Isobutyldiol-POSS as chain extender to react with 4,4′ diphenyl methylene diisocyanate. -

-
Scheme 3 shows an example of the synthesis of TPU using a polycyclooctene diol as polyol (n is chosen such that the diol molecular weight is in the range of about 2,000 to about 20,000 g/mol), TMP Isobutyldiol-POSS as chain extender to react with 4,4′ diphenyl methylene diisocyanate. -

-
Scheme 4 illustrates an example of the synthesis of polycaprolactone-polylactide random copolymer diol using ring-opening polymerization. The copolymers show one single sharp Tg that can be tunable according to the CL:LA molar ratios. In an exemplary embodiment, D,L-lactide or “meso” lactide (CAS # 96-95-5,3,6-dimethyl-1,4-dioxane-2,5-dione) is reacted with caprolactone to provide the copolymer. -

- Scheme 5 shows an example of synthesis of TPU using a polycaprolactone-D,L-polylactide random copolymer as polyol, TMP Isobutyldiol-POSS(R is isobutyl) as chain extender to react with 4,4′ diphenyl methylene diisocyanate.
- A general structure for the POSS-based TPUs incorporating PEG diol and TMP POSS is shown in
Scheme 6. The polymers allow systematic variation in the ratio of x/y (1 to 20), the polyol degree of polymerization (1≦n≦1000), and the total degree of polymerization, 2≦m≦100. -
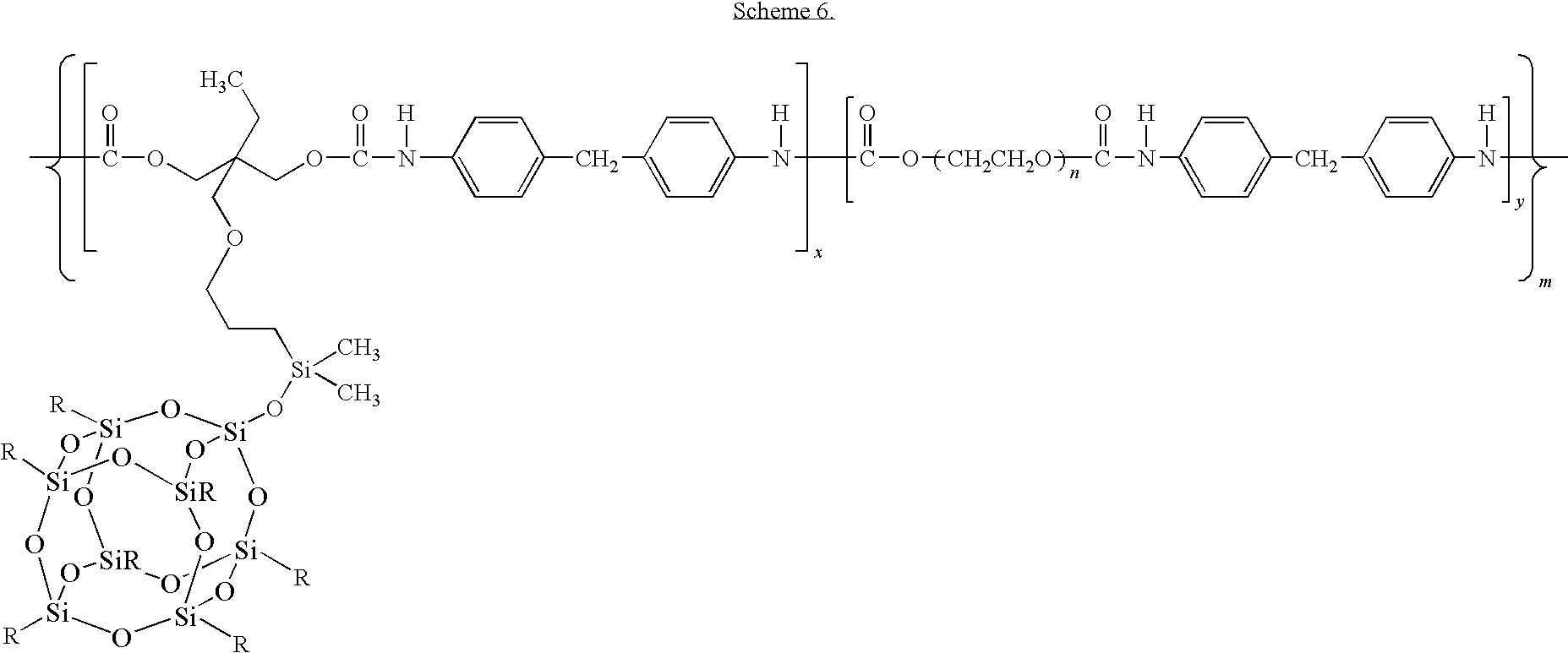
- In the foregoing scheme, the R substituent can include a C1-C12 primary, secondary, or tertiary alkyl group. Exemplary R groups include methyl, isobutyl, isooctyl, cyclopentyl, cyclohexyl, phenyl, and the like.
- The instant hybrid polyurethanes demonstrate sharp and tunable transition temperatures, adjustable stiffness above their transition temperatures, and thermal processability above the melting point of the POSS domains. The hybrid polyurethanes also show excellent shape recovery effect at the recovery temperature and the retracting force is adjustable according to the composition of the POSS. They also posses a unique property that is different from the other shape memory polymers in that the current invention (in the PEG embodiment) can be triggered to recover by moisture (liquid or vapor) aside from heating. For the thermal triggering mechanism, the
range 30° C. to 60° C. according to the ratio of the components used and (importantly) thermal annealing to achieve steady-state (equilibrium) crystallinity is important. The recovery can be finished within seconds when heated 20° C. above the transition temperature. The additional advantages of the materials include that the materials are rigid at room temperature, the polymers generally are biocompatible. In some cases, the described TPUs are biodegradable, and the biodegradation rate can be controlled by the chemical compositions, soft segment length, and soft-segment/hard-segment ratios, and can be used as medical devices and implants, as the degradation products are generally non-toxic, non-immunogenic, and absorbable. Biodegradability can be estimated by measuring the percent mass loss after 70 days immersion in a buffered saline solution (0.01 M phosphate, 0.138 M NaCl, 0.027 M KCl). This test is described in detail in the working examples below. Some samples exhibit less than about 85% mass loss in this test, while others exhibit greater than about 85% mass loss. The materials can also be used as drug elution stents or stent coatings. The materials can further be dyed to any color or rendered radio-opaque for x-ray radiography according to application requirements. - Any of the hybrid polyurethane polymers mentioned above may be filled with, for example, nanoparticles of boron nitride, silica, titanium dioxide, montmorillonite, clay, Kevlar, staple, aluminum nitride, barium subcarbonate and bismuth subcarbonate. Clay and silica can be used to, for example, increase the modulus of the plastic. Dispersing agents and/or compatibilizing agents may be used, for example, to improve the blending of polymers and the blending of polymers with fillers. Dispersing agents and/or compatibilizing agents include, for example, ACRAWAX® (ethylene bis-stearamide), polyurethanes and ELVALOY® (acrylic functionalized polyethylene).
- In one embodiment, the rate of biodegradation of the thermoplastic polyurethane shape memory polymer can be controlled by adjusting the content of POSS chain extender, the content and composition of the polyol. For example, the biodegradation rate can be a) decreased by increasing the amount of POSS chain extender in the polymer; b) decreased by increasing the molecular weight of the polyol in the polymer; or c) increased by increasing the amount of hydrolysable groups in the polyol.
- In another embodiment, a drug eluting implant, a drug eluting stent, or drug eluting stent coating is prepared from a biodegradable shape memory polymer. For example, a thermoplastic polyurethane shape memory polymer can exhibit a certain rate of biodegradation. The rate of drug elution from the polymer correlates to the rate of biodegradation of the shape memory polymer.
-
FIG. 1 illustrates graphically the DMA plots of the TMP POSS based thermoplastic polyurethane (TPU) with mole ratio of PEG:POSS as 1:6, 1:4 respectively; -
FIG. 2 illustrates graphically the DSC results of TMP POSS based TPU with different PEG:POSS mole ratios; -
FIG. 3 illustrates visually the response of an example TPU (TMP POSS based TPU (PEG:POSS=1:6)) to a large tensile deformation; -
FIG. 4 illustrates graphically the stress-strain plot of the TMP POSS based TPU (PEG:POSS=1:6); -
FIG. 5 illustrates graphically the mass loss of TPUs with different soft segment length and soft-segment/hard-segment ratios in phosphate buffered saline; -
FIG. 6 illustrates graphically the molecular weight decrease of TPUs with different soft segment length and soft-segment/hard-segment ratios in phosphate buffered saline; and -
FIG. 7 illustrates graphically the critical temperature change of a TPU in phosphate buffered saline. - Thermoplastic polyurethanes with different compositions were synthesized by one-step condensation
polymerization using scheme 1 shown above ((A) PEG as the polyol, (B) transcyclohexanediolisobutyl-polyhedral oligosilsesquioxane diol and (C) MDI as the diisocyanate. Toluene was used as solvent and dibutyltin dilaurate was used as catalyst. The reaction was kept at 90° C. under the nitrogen for 2 hours and then cooled down to room temperature and precipitated into hexane. The product was dried thoroughly and dissolved in toluene to make a 10 wt % solution for casting films. The molecular weights and molecular weight distributions of this series of samples obtained from size exclusion chromatography are summarized in Table 1. -
TABLE 1 Molecular weights and molecular weight distributions of POSS-based polyurethanes having polyol (PEG) block length of 10000 g/mol Sample Mn (g/mol) Mw/Mn PEG:POSS = 47,400 1.42 1:3 PEG:POSS = 48,800 1.44 1:4 PEG:POSS = 54,000 1.54 1:6 PEG:POSS = 49,200 1.30 1:8 - Samples of polyurethanes with different compositions were characterized by differential scanning calorimetry (TA Instruments DSC2920). All of the samples were characterized under the same conditions: two scans were performed for each sample with heating and cooling rates of 10° C./min (
FIG. 2 ). It was observed that this series of polyurethanes exhibit two melting points, one in the range 45<Tm1, <50° C. corresponding to the melting temperature of PEG “soft” block. The other melting transition appears in the range 110<Tm2<130° C., which corresponds to the melting of a POSS-reinforced hard segment phase. The melting temperature of the soft segment is observed to shift to lower values with a broadening of the melting peak while the melting temperature of the hard segment is observed to shift to higher values with a sharpening of the melting peak when the mole ratio of polyol:chain extender decreases. This result can be explained in that as the PEG:POSS ratio decreases, the resulting block copolymer will have less overall PEG content, which will directly affect the size and perfection of the crystallization of PEG blocks. Therefore, the melting temperature moves to lower values and the peak is broadened. On the contrary, the content of POSS will increase in the block copolymers, which provides for more clear aggregation of hard segments to form larger and more perfect crystals. Therefore, the melting temperature of hard segment moves to higher values while the peak is sharpened (FIG. 2 ). - The dried films of the formed polyurethanes were cut into thin strips for tests of temporary shape fixing and subsequent recovery, or shape memory. For example, a sample was first heated on the hot stage to 65° C., which is well above the first transition temperature but low enough to avoid melting of the elastic network of the POSS-rich phase. It was then stretched to a certain degree of elongation and cooled down to the room temperature. The deformed shape was fixed at room temperature. Finally, the deformed sample was heated up again on hot plate to 65° C. and it was observed that the sample restored to its original length completely and within seconds. A similar phenomenon was observed when water was used as a stimulus for the shape recovery except that the sample secondarily swelled to form a tough hydrogel.
- Biodegradation studies were performed on polyurethanes prepared from poly(ε-caprolactone)-co-poly(D,L-lactide) as the polyol; 1-[2-ethyl-2-[(3-dimethylsiloxy)propoxymethyl]-1,3-propanediol]-3,5,7,9,11,13,15-isobutylpentacyclo-[9.5.113,9.15,15.17,13]-octasiloxane as the dihydroxyl-terminated POSS; and 4,4-methylenebis(phenyl-isocyanate) as the diisocyanate according to the following procedure.
- Synthesis of poly(ε-caprolactone)-co-poly(D,L-lactide) (PCL-co-PDLLA) was carried out in bulk. A mixture of ε-caprolactone (99%, Aldrich, used as received), D, L-lactide (99%, Aldrich, purified by recrystallization), 1,4-butane diol (96%, Aldrich, dried using 4 Å molecular sieves) (molar amounts varied) and catalytic stannous octoate were put in a three-neck flask equipped with nitrogen inlet and outlet. Magnetic stirring was used to agitate the reaction mixture. The temperature was set at 140° C. and the polymerization reaction took 4 hours to complete. The product was dissolved in toluene and precipitated into hexane. The sample of PCL-co-PDLLA was dried in vacuum oven overnight.
- POSS-based thermoplastic polyurethanes were then synthesized using a one-step polymerization method. A 50 mL three-neck flask equipped with nitrogen inlet and outlet, condenser and thermometer was used as the reactor for the polymerization. Starting with a purging of nitrogen, 1.5 g of PCL-co-PDLLA (Mn=10000 g/mol, 1.5×10−4 mol) were mixed together with 1 g (9.03×10−4 mol) of TMP-DiolIsobutyl-POSS (1-[2-Ethyl-2-[(3-dimethylsiloxy)propoxymethyl]-1,3-propanediol]-3,5,7,9,11,13,15-isobutylpentacyclo-[9.5.113,9.15,15.17,13]-octasiloxane, 95%, Hybrid Plastics, used as received) in 25 mL of toluene (Fisher, ACS Certified, dried over molecular 4 Å sieves). The reactor was heated to 50° C., followed by adding 0.2633 g (1.05×10−3 mol) of 4,4-methylenebis(phenyl-isocyanate) (98%, Aldrich, used as received). The reaction mixture was then heated to 90° C. and 2 drops of dibutyltin dilaurate (95%, Aldrich) was added through a syringe. A thickening phenomenon was observed during the reaction and the reaction was kept at 90° C. for 2 hr under the nitrogen atmosphere before completion. Then the thickened polymer solution was precipitated into excess amount of n-hexane, filtered and dried to remove any unreacted POSS. The dried polymer was dissolved in toluene again to make a 10 wt % of solution for casting films. Several series of samples were made using PCL-co-PDLLA as soft segment and the ratio of PCL-co-PDLLA/POSS as variables (See
FIG. 5 ). - Biodegradation test of samples (approximately 150 micrometers thick) with different polyol molecular weight and different polyol:POSS ratio was carried out at 37° C. in Phosphate Buffered Saline (PBS) ((0.01 M); NaCl 0.138 M; KCl 0.0027 M) buffer. The samples were put in the buffer for 92 days and the progress of degradation was measured every 10 days by taking the samples from the buffer and determining the percent mass retained. The data are summarized in
FIG. 5 . - As illustrated by
FIG. 5 , it was observed that the degradation was very slow at the first 30 days. The samples having higher molecular weight of soft segment degraded more slowly than the ones having low molecular weight of soft segment. Moreover, with same molecular weight of soft segment, those having higher POSS content degraded more slowly than the ones having lower POSS content. Therefore, it can be speculated that the soft segment is a dominant species at the beginning of the degradation. The degradation progressed by chain scission of the soft segment, particularly hydrolysis of the ester linkages (Schemes 4 and 5). Therefore, the longer the soft segment, the slower the rate of degradation. Moreover, if the weight fraction of the POSS is higher, the same chain scission is suppressed and the rate of degradation will also be decreased. - After the first 30 days, the rate of degradation was accelerated for most of the samples, with those having lower molecular weight of soft segment having a higher rate of degradation. After 50 days of degradation, cracks appeared on the sample surface for those having lower soft segment molecular weight. The cracks continued to grow until the top layer of the surface separated. With continued degradation, this phenomenon was observed for all samples sooner or later. The profile of the mass loss of the samples is shown in
FIG. 5 . - Size Exclusion Chromatography (SEC) of samples degraded as above for 0, 8, 60, and 82 days was conducted for SMP polyurethane samples having 20 kDa soft segment, polyol:POSS=1.8 as well as 35.5 kDa soft segment, polyol:POSS=1:15 (See
FIG. 6 ). It was observed that although the mass loss is very slow at the beginning of the degradation, the MW during the same stage decreased very fast further proving that the degradation proceeds by chain scission. After the sudden decrease in MW within the first 8 days, the molecular weight decreased slowly during the remaining time. - Thermal analysis by differential scanning calorimetry (DSC) of selected samples at 0, 8, 60, and 82 days was also conducted (see
FIG. 7 ). All of the samples were first heated to 160° C. at 20° C./min, then cooled down to 70° C. and annealed for 30 min. After annealing, they were cooled down to −20° C. and equilibrated at that temperature and isothermal for 1 min. This preconditioning was conducted in order to achieve a reproducible state comparable between samples. Finally, they were heated to 160° C. again at 20° C./min. and the heat flows were recorded (SeenFIG. 7 ). It was observed that with ongoing degradation, the Tg of the sample was broadened and the Tm shifted to higher temperature with the appearance of multi-melting peaks. - The hybrid polyurethanes of the invention can be used for the following applications.
-
- a. Stents, patches and other implants for human health care
- b. Surgical tools requiring adjustable shape but high stiffness.
- c. Arbitrarily shape-adjustable structural implements, including personal care items (dinnerware, brushes, etc.) and hardware tool handles.
- d. Self healing plastics
- e. Medical devices (a dented panel is repaired by heating or plasticizing with solvent)
- f. Drug delivery matrices
- g. High-strength thermoplastic (non-crosslinked) superabsorbant hydrogels
- h. Aqueous rheological modifiers for paints, detergents, and personal care products
- i. Impression material for molding, duplication, rapid prototyping, dentistry, and figure-printing.
- j. Toys
- k. Reversible Embossing for information storage
- l. Temperature and moisture sensors
- m. Safety valve
- n. Heat shrink tapes or seals
- o. Heat controlled Couplings and fasteners
- p. Large strain, large force actuators
- q. Coatings, adhesives
- r. Textiles, clothing
- The shape memory polymers of the disclosure are particularly suitable as biomaterials because of their low thrombogenicity, high biocompatibility, as well as unique mechanical properties. In accordance with the invention the shape memory polyurethanes were formulated such that the melting temperature of one segment falls within a useful temperature range for biomedical application: 37° C.-50° C.
- The present disclosure provides an advantageous shape memory polymer that includes thermoplastic polyurethane shape memory polymers formed by reacting in one step a polyol, a POSS chain extender and a diisocyanate, having medium and tunable modulus in the fixed state at room temperature having a tunable sharp transition, whose permanent shape can be repeatedly remolded above a certain melting temperature.
- The terms “a” and “an” herein do not denote a limitation of quantity, but rather denote the presence of at least one of the referenced item. All ranges disclosed herein are inclusive and combinable. All cited patents, patent applications, and other references are incorporated herein by reference in their entirety.
- Although the polymers and processing methodologies of the present disclosure have been described with reference to specific exemplary embodiments thereof, the present disclosure is not to be limited to such exemplary embodiments. Rather, as will be readily apparent to persons skilled in the art, the teachings of the present disclosure are susceptible to many implementations and/or applications, without departing from either the spirit or the scope of the present disclosure. Indeed, modifications and/or changes in the selection of specific polymers, polymer ratios, processing conditions, and end-use applications are contemplated hereby, and such modifications and/or changes are encompassed within the scope of the present invention as set forth in the claims which follow.
Claims (22)
1-19. (canceled)
20. A method for making a thermoplastic polyurethane shape memory polymer comprising reacting (A) a polyol, (B) a polyhedral oligosilsesquioxane diol, and (C) a diisocyanate; wherein the shape memory polymer exhibits a thermal triggering temperature of 30° C. to 60° C.
21. The method of claim 20 wherein the polyhedral oligosilsesquioxane diol is a member selected from the group consisting of 2-ethyl-2-[3-[[(heptacyclopentylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxanyl)oxy]dimethylsilyl]-propoxy]methyl]-1,3-propanediol, 2-ethyl-2-[3-[[(heptacyclohexylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxanyl)oxy]dimethylsilyl]-propoxy]methyl]-1,3-propanediol, 2-ethyl-2-[3-[[(heptaisobutylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxanyl)oxy]dimethylsilyl]-propoxy]methyl]-1,3-propanediol, 1-(2-trans-cyclohexanediol)ethyl-3,5,7,9,11,13,15-cyclohexanepentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxane, and 1-(2-trans-cyclohexanediol)ethyl-3,5,7,9,11,13,15-isobutylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxane.
22. The method of claim 20 wherein the diisocyanate is a member selected from the group consisting of 4,4′-diphenyl methylene diisocyanate (MDI), toluene-2,4-diisocyanate (TDI), toluene-2,6-diisocyanate, hexamethylene-1,6-diisocyanate (HDI), isophorone diisocyanate (IPDI), and hydrogenated 4,4′-diphenylmethane diisocyanate (H12MDI).
23. The method of claim 20 wherein the diisocyanate is 4,4′-diphenyl methylene diisocyanate.
24. The method of claim 20 , wherein the thermoplastic polyurethane shape memory polymer exhibits a thermal triggering temperature of 37° C. to 50° C.
25. The method of claim 20 , wherein the polyol is a member selected from the group consisting of polyethylene glycol (PEG), polycaprolactone (PCL) diol, polycyclooctene diol, polynorbornene diol and polymethacrylate copolymer.
26. The method of claim 20 , wherein the polyol is a member selected from the group consisting of polyethylene glycol, polycaprolactone diol, and polycyclooctene diol, and is semicrystalline.
27. The method of claim 20 , wherein the polyol is an amorphous diol having a Tg in the range of 20 to 80° C., and is a member selected from the group consisting of polynorbornene diol and polymethacrylate copolymer diol.
28. A method for making a thermoplastic polyurethane polymer comprising reacting (A) a polyol, (B) a polyhedral oligosilsesquioxane diol, and (C) a diisocyanate, wherein the polyol is a member selected from the group consisting of polyethylene glycol, polycaprolactone diol, and polycyclooctene diol; wherein the polyhedral oligosilsesquioxane diol is a member selected from the group consisting of 2-ethyl-2-[3-[[(heptacyclopentylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxanyl)oxy]dimethylsilyl]-propoxy]methyl]-1,3-propanediol, 2-ethyl-2-[3-[[(heptacyclohexylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxanyl)oxy]dimethylsilyl]-propoxy]methyl]-1,3-propanediol, 2-ethyl-2-[3-[[(heptaisobutylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxanyl)oxy]dimethylsilyl]-propoxy]methyl]-1,3-propanediol, 1-(2-trans-cyclohexanediol)ethyl-3,5,7,9,11,13,15-cyclohexanepentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxane, and 1-(2-trans-cyclohexanediol)ethyl-3,5,7,9,11,13,15-isobutylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxane; and wherein the diisocyanate is 4,4′-diphenyl methylene diisocyanate.
29. The method of claim 28 wherein said reaction is carried out in presence of dibutyltin dilaurate as catalyst.
30. The method of claim 20 , wherein the polyol is polyethylene glycol, wherein the polyhedral oligosilsesquioxane is 1-(2-trans-cyclohexanediol)ethyl-3,5,7,9,11,13,15-isobutylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxane, and wherein the diisocyanate is 4,4′-diphenyl methylene diisocyanate.
31. The method of claim 20 , wherein the polyol is polycyclooctene diol, wherein the polyhedral oligosilsesquioxane diol is 2-ethyl-2-[3-[[(heptaisobutylpentacyclo-[9.5.1.13,9.15,15.17,13]octasiloxanyl)oxy]dimethylsilyl]-propoxy]methyl]-1,3-propanediol, and wherein the diisocyanate is 4,4′-diphenyl methylene diisocyanate.
32. A thermoplastic polyurethane polymer prepared by the method of claim 20 .
33. A thermoplastic polyurethane polymer prepared by the method of claim 28 .
34. A thermoplastic polyurethane polymer prepared by the method of claim 30 .
35. A thermoplastic polyurethane polymer prepared by the method of claim 31 .
36. A composition comprising
a thermoplastic polyurethane shape memory polymer prepared according to the method of claim 20 , and
a filler selected from the group consisting of boron nitride, silica, titanium dioxide, montmorillonite, clay, staple, aluminum nitride, barium subcarbonate, and bismuth subcarbonate.
37. The thermoplastic polyurethane shape memory polymer of claim 36 , wherein the thermoplastic shape memory polymer exhibits a thermal triggering temperature of 37° C. to 50° C.
38. A thermoplastic polyurethane polymer having the formula

wherein R is an alkyl group having six or fewer carbon atoms; wherein the ratio of x to y is 1:1 to 20:1; wherein the polyol degree of polymerization, n, is 1 to 1000; and wherein the total degree of polymerization, m, is 2 to 100.
39. The thermoplastic polyurethane polymer of claim 38 , wherein R is an alkyl group having 4 to 6 carbon atoms.
40. The thermoplastic polyurethane polymer of claim 38 , wherein R is isobutyl, cyclopentyl, or cyclohexyl.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/403,410 US20090253842A1 (en) | 2002-10-11 | 2009-03-13 | Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US41802302P | 2002-10-11 | 2002-10-11 | |
| US41950602P | 2002-10-18 | 2002-10-18 | |
| US46640103P | 2003-04-29 | 2003-04-29 | |
| US48859003P | 2003-07-18 | 2003-07-18 | |
| US48832303P | 2003-07-18 | 2003-07-18 | |
| US10/683,167 US7091297B2 (en) | 2002-10-11 | 2003-10-10 | Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments |
| US11/111,388 US7524914B2 (en) | 2002-10-11 | 2005-04-21 | Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments |
| US12/403,410 US20090253842A1 (en) | 2002-10-11 | 2009-03-13 | Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/111,388 Continuation US7524914B2 (en) | 2002-10-11 | 2005-04-21 | Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20090253842A1 true US20090253842A1 (en) | 2009-10-08 |
Family
ID=36677269
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/111,388 Expired - Fee Related US7524914B2 (en) | 2002-10-11 | 2005-04-21 | Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments |
| US12/403,410 Abandoned US20090253842A1 (en) | 2002-10-11 | 2009-03-13 | Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/111,388 Expired - Fee Related US7524914B2 (en) | 2002-10-11 | 2005-04-21 | Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US7524914B2 (en) |
| EP (1) | EP1907434A1 (en) |
| JP (1) | JP2008537010A (en) |
| AU (1) | AU2006240293B2 (en) |
| CA (1) | CA2605497A1 (en) |
| WO (1) | WO2006115799A1 (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090042999A1 (en) * | 2007-08-09 | 2009-02-12 | Samsung Electronics Co., Ltd. | Composition for polyurethane foam, polyurethane foam made from the composition, and method for preparing polyurethane foam |
| US20110092652A1 (en) * | 2009-10-20 | 2011-04-21 | Voit Walter E | Shape memory polymers and process for preparing |
| DE102010040762A1 (en) * | 2010-09-14 | 2012-03-15 | Technische Hochschule Wildau | Inorganic-organic polymer hybrid network, useful for preparing shape memory polymer, comprises hydroxyl group-containing thermoplastically processable polymer, nanoscale metal compounds, and reactive compounds |
| CN102617823A (en) * | 2012-03-29 | 2012-08-01 | 合肥工业大学 | Process for preparing hydroxyl polyhedral oligomeric silsesquioxane modified polyurethane |
| US8815054B2 (en) * | 2012-10-05 | 2014-08-26 | The Procter & Gamble Company | Methods for making fibrous paper structures utilizing waterborne shape memory polymers |
| CN109867947A (en) * | 2017-12-01 | 2019-06-11 | 财团法人纺织产业综合研究所 | Particles and yarn |
| US10407574B2 (en) * | 2016-10-20 | 2019-09-10 | Samsung Electronics Co., Ltd. | Composition with self-healing property, film with self-healing property and device including the film |
| US10563004B2 (en) | 2015-03-27 | 2020-02-18 | Basf Se | Memory foam based on thermoplastic polyurethane |
| CN111690111A (en) * | 2020-07-30 | 2020-09-22 | 中国科学院兰州化学物理研究所 | Comb type polymer and preparation method and application thereof |
Families Citing this family (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040030062A1 (en) * | 2002-05-02 | 2004-02-12 | Mather Patrick T. | Castable shape memory polymers |
| US7794494B2 (en) | 2002-10-11 | 2010-09-14 | Boston Scientific Scimed, Inc. | Implantable medical devices |
| US7976936B2 (en) | 2002-10-11 | 2011-07-12 | University Of Connecticut | Endoprostheses |
| ATE534704T1 (en) * | 2002-10-11 | 2011-12-15 | Univ Connecticut | CROSS-LINKED POLYCYCLOOCTENE |
| CA2501549C (en) * | 2002-10-11 | 2011-08-30 | University Of Connecticut | Blends of amorphous and semicrystalline polymers having shape memory properties |
| US7931693B2 (en) | 2004-02-26 | 2011-04-26 | Endosphere, Inc. | Method and apparatus for reducing obesity |
| US8585771B2 (en) | 2004-02-26 | 2013-11-19 | Endosphere, Inc. | Methods and devices to curb appetite and/or to reduce food intake |
| US8147561B2 (en) | 2004-02-26 | 2012-04-03 | Endosphere, Inc. | Methods and devices to curb appetite and/or reduce food intake |
| CA2593471A1 (en) * | 2004-12-10 | 2006-07-06 | University Of Connecticut | Shape memory polymer orthodontic appliances, and methods of making and using the same |
| US9060835B2 (en) | 2006-05-26 | 2015-06-23 | Endosphere, Inc. | Conformationally-stabilized intraluminal device for medical applications |
| US20080085946A1 (en) * | 2006-08-14 | 2008-04-10 | Mather Patrick T | Photo-tailored shape memory article, method, and composition |
| US20080132988A1 (en) * | 2006-12-01 | 2008-06-05 | Scimed Life Systems, Inc. | Balloon geometry for delivery and deployment of shape memory polymer stent with flares |
| US8684101B2 (en) * | 2007-01-25 | 2014-04-01 | Tini Alloy Company | Frangible shape memory alloy fire sprinkler valve actuator |
| US8584767B2 (en) * | 2007-01-25 | 2013-11-19 | Tini Alloy Company | Sprinkler valve with active actuation |
| US8617237B2 (en) * | 2007-02-16 | 2013-12-31 | Universität Zürich | Tubular supporting prosthesis with a heart valve, in particular for aortic valve replacement |
| CA2721855C (en) * | 2007-04-19 | 2017-05-30 | University Of Massachusetts Medical School | Thermal-responsive polymer siloxanes, compositions, and methods and applications related thereto |
| US20110137227A1 (en) | 2007-07-16 | 2011-06-09 | Mckinley James T | Methods and devices for delivering or delaying lipids within a duodenum |
| WO2009018289A2 (en) * | 2007-07-30 | 2009-02-05 | Tini Alloy Company | Method and devices for preventing restenosis in cardiovascular stents |
| US20090035350A1 (en) | 2007-08-03 | 2009-02-05 | John Stankus | Polymers for implantable devices exhibiting shape-memory effects |
| US9259515B2 (en) | 2008-04-10 | 2016-02-16 | Abbott Cardiovascular Systems Inc. | Implantable medical devices fabricated from polyurethanes with grafted radiopaque groups |
| US8136536B2 (en) * | 2008-09-24 | 2012-03-20 | Elc Management Llc | Shape memory polymer mascara brush |
| EP2349080B1 (en) | 2008-10-22 | 2016-04-13 | Boston Scientific Scimed, Inc. | Shape memory tubular stent with grooves |
| US9533469B2 (en) * | 2008-12-23 | 2017-01-03 | Syracuse University | Self-healing product |
| US8633292B2 (en) * | 2009-03-26 | 2014-01-21 | Signet Armorlite | Polyurethane-based photochromic optical materials |
| US8404484B2 (en) * | 2009-07-15 | 2013-03-26 | Syracuse University | Active cell culture via shape memory |
| BR112012013200B1 (en) * | 2009-12-21 | 2019-08-13 | Huntsman Int Llc | method for forming a urethane material, reactive composition, and polyurethane material |
| US8683798B2 (en) * | 2010-01-15 | 2014-04-01 | Syracuse University | Stimuli-responsive product |
| SG184102A1 (en) * | 2010-03-18 | 2012-10-30 | Agency Science Tech & Res | Biodegradable and biocompatible shape memory polymers |
| TWI537314B (en) * | 2010-04-08 | 2016-06-11 | 國立清華大學 | Micro-fluidic power system and a method to produce the same |
| US9249254B2 (en) * | 2010-04-22 | 2016-02-02 | Syracuse University | Polyhedral oligomeric silsesquioxane polyurethanes |
| US9180228B2 (en) * | 2011-10-03 | 2015-11-10 | Abbott Cardiovascular Systems Inc. | Rubber toughened bioresorbable polymer peripheral scaffolds |
| CN102408539A (en) * | 2011-10-24 | 2012-04-11 | 哈尔滨工业大学 | Shape memory polyurethane and preparation method thereof |
| JP2013223574A (en) * | 2012-04-20 | 2013-10-31 | Olympus Medical Systems Corp | Elastomer molding for medical instrument |
| EP2864386A1 (en) * | 2012-06-25 | 2015-04-29 | Lubrizol Advanced Materials, Inc. | Method for identifying bioabsorbable polymers |
| WO2014004334A1 (en) * | 2012-06-25 | 2014-01-03 | Lubrizol Advanced Materials, Inc. | Process for making biodegradable and/or bioabsorbable polymers |
| US11040230B2 (en) | 2012-08-31 | 2021-06-22 | Tini Alloy Company | Fire sprinkler valve actuator |
| US10124197B2 (en) | 2012-08-31 | 2018-11-13 | TiNi Allot Company | Fire sprinkler valve actuator |
| US9943298B2 (en) | 2012-10-19 | 2018-04-17 | Cook Medical Technologies Llc | Vascular closure with shape memory characteristic |
| US9422393B2 (en) * | 2012-11-13 | 2016-08-23 | Syracuse University | Water-triggered shape memory of PCL-PEG multiblock TPUs |
| CA2950662C (en) * | 2014-05-30 | 2021-11-02 | Stuart Arthur Bateman | Ice adhesion reducing prepolymers and polymers |
| US10448678B2 (en) | 2014-08-13 | 2019-10-22 | Mast Industries (Far East) Limited | Bra incorporating shape memory polymers and method of manufacture thereof |
| EP3061777B1 (en) | 2015-02-24 | 2021-11-24 | Albert-Ludwigs-Universität Freiburg | Phase-segregated block copolymers with tunable properties |
| CN108473651B (en) | 2015-12-22 | 2022-01-25 | 巴斯夫欧洲公司 | TPU shrink material |
| CN109661306A (en) * | 2016-09-12 | 2019-04-19 | 科思创德国股份有限公司 | Low temperature increasing material manufacturing method based on fusion sediment model |
| JP6919992B2 (en) * | 2017-01-30 | 2021-08-18 | 信越化学工業株式会社 | Elastic film and its formation method |
| KR101963781B1 (en) * | 2017-09-20 | 2019-07-31 | 포항공과대학교 산학협력단 | Shape-Memory Polymer Sheet for Fabrication of Shape-Specific Cell Sheet |
| US10430118B2 (en) * | 2017-10-27 | 2019-10-01 | Ncr Corporation | Transparent device driver integration |
| JP7132133B2 (en) * | 2018-02-08 | 2022-09-06 | 信越化学工業株式会社 | Stretchable membrane material composition, stretchable membrane, and method for forming the same |
| JP6978375B2 (en) * | 2018-04-23 | 2021-12-08 | 信越化学工業株式会社 | Adhesive film, method of forming adhesive film, and urethane polymer |
| CN108559054B (en) * | 2018-05-02 | 2021-02-02 | 国家纳米科学中心 | Shape memory polymer and preparation method and application thereof |
| EP3594257A1 (en) | 2018-07-13 | 2020-01-15 | Albert-Ludwigs-Universität Freiburg | Use of phase segregated block copolymers with tiunable properties for the coating or surfces and coated substrates |
| CN109575221A (en) * | 2018-12-04 | 2019-04-05 | 镇江利德尔复合材料有限公司 | A kind of polyurethane shape memory material and preparation method thereof strengthened based on organosilicon polymer |
| CN109575570A (en) * | 2018-12-06 | 2019-04-05 | 镇江利德尔复合材料有限公司 | A kind of preparation method of the excellent polyurethane material of novel shape-memory properties |
| CN111269373B (en) * | 2020-02-12 | 2022-01-04 | 浙江大学衢州研究院 | Preparation method of remodelable shape memory elastomer based on eutectic |
| CN113248741B (en) * | 2021-05-20 | 2022-11-11 | 深圳市银宝山新科技股份有限公司 | Preparation method of cross-linked shape memory polyurethane |
| CN114276655B (en) * | 2021-07-29 | 2023-04-25 | 南京工程学院 | Degradable thermoplastic elastomer and preparation method thereof |
| CN114891180B (en) * | 2022-05-16 | 2023-06-27 | 北京化工大学 | Self-repairable siloxane modified polyurethane material and preparation method thereof |
| CN115154671B (en) * | 2022-07-15 | 2023-06-16 | 重庆大学 | Polylactic acid and shape memory polyurethane material compound |
Citations (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3383336A (en) * | 1966-06-27 | 1968-05-14 | Kuyama Hiroshi | Resinous insoluble reaction products |
| US3459725A (en) * | 1964-01-17 | 1969-08-05 | Montedison Spa | High molecular weight unsaturated hydrocarbon homopolymers and process for preparing them |
| US3563973A (en) * | 1965-03-04 | 1971-02-16 | Radiation Applic Inc | Articles with polymeric memory and method of constructing same |
| US4612241A (en) * | 1985-06-12 | 1986-09-16 | E. I. Du Pont De Nemours And Company | Impact resistant composites with elastomeric fibers |
| US5145935A (en) * | 1988-09-30 | 1992-09-08 | Mitsubishi Jukogyo Kabushiki Kaisha | Shape memory polyurethane elastomer molded article |
| US5147385A (en) * | 1989-11-01 | 1992-09-15 | Schneider (Europe) A.G. | Stent and catheter for the introduction of the stent |
| US5163952A (en) * | 1990-09-14 | 1992-11-17 | Michael Froix | Expandable polymeric stent with memory and delivery apparatus and method |
| US5189110A (en) * | 1988-12-23 | 1993-02-23 | Asahi Kasei Kogyo Kabushiki Kaisha | Shape memory polymer resin, composition and the shape memorizing molded product thereof |
| US5258020A (en) * | 1990-09-14 | 1993-11-02 | Michael Froix | Method of using expandable polymeric stent with memory |
| US5282854A (en) * | 1989-10-11 | 1994-02-01 | Daikin Industries Ltd. | Fluorine-containing block copolymer and artificial lens comprising the same |
| US5395882A (en) * | 1992-09-24 | 1995-03-07 | Rohm Gmbh Chemische Fabrik | Light-scattering polystyrene molding compounds, and molded articles produced therefrom |
| US5506300A (en) * | 1985-01-04 | 1996-04-09 | Thoratec Laboratories Corporation | Compositions that soften at predetermined temperatures and the method of making same |
| US5665822A (en) * | 1991-10-07 | 1997-09-09 | Landec Corporation | Thermoplastic Elastomers |
| US5674242A (en) * | 1995-06-06 | 1997-10-07 | Quanam Medical Corporation | Endoprosthetic device with therapeutic compound |
| US5769883A (en) * | 1991-10-04 | 1998-06-23 | Scimed Life Systems, Inc. | Biodegradable drug delivery vascular stent |
| US5880240A (en) * | 1996-04-03 | 1999-03-09 | Arakawa Chemical Industries, Ltd. | Alkyl-containing porous resin, process for its preparation and its use |
| US5889118A (en) * | 1996-06-03 | 1999-03-30 | Minnesota Mining And Manufacturing Company | Thermomorphic "smart" pressure sensitive adhesives |
| US5900444A (en) * | 1996-10-08 | 1999-05-04 | Zamore; Alan | Irradiation conversion of thermoplastic to thermoset polyurethane |
| US5908918A (en) * | 1996-07-17 | 1999-06-01 | Chronopol, Inc. | Impact modified polylactide |
| US5910357A (en) * | 1996-07-12 | 1999-06-08 | Nitto Denko Corporation | Separation membrane and method of producing the same, and shape memory polymer composition |
| US5954744A (en) * | 1995-06-06 | 1999-09-21 | Quanam Medical Corporation | Intravascular stent |
| US5955559A (en) * | 1996-09-17 | 1999-09-21 | Shell Oil Company | Cast polyurethane elastomers containing low polarity amine curing agents |
| US5964744A (en) * | 1993-01-04 | 1999-10-12 | Menlo Care, Inc. | Polymeric medical device systems having shape memory |
| US6024764A (en) * | 1997-08-19 | 2000-02-15 | Intermedics, Inc. | Apparatus for imparting physician-determined shapes to implantable tubular devices |
| US6086204A (en) * | 1999-09-20 | 2000-07-11 | Magnante; Peter C. | Methods and devices to design and fabricate surfaces on contact lenses and on corneal tissue that correct the eye's optical aberrations |
| US6156842A (en) * | 1998-03-11 | 2000-12-05 | The Dow Chemical Company | Structures and fabricated articles having shape memory made from α-olefin/vinyl or vinylidene aromatic and/or hindered aliphatic vinyl or vinylidene interpolymers |
| US6160084A (en) * | 1998-02-23 | 2000-12-12 | Massachusetts Institute Of Technology | Biodegradable shape memory polymers |
| US6183248B1 (en) * | 1998-11-30 | 2001-02-06 | Muhammad Chishti | System and method for releasing tooth positioning appliances |
| US6217609B1 (en) * | 1998-06-30 | 2001-04-17 | Schneider (Usa) Inc | Implantable endoprosthesis with patterned terminated ends and methods for making same |
| US6248129B1 (en) * | 1990-09-14 | 2001-06-19 | Quanam Medical Corporation | Expandable polymeric stent with memory and delivery apparatus and method |
| US20020007222A1 (en) * | 2000-04-11 | 2002-01-17 | Ashvin Desai | Method and apparatus for supporting a body organ |
| US20020015519A1 (en) * | 1998-12-11 | 2002-02-07 | Lord Corporation | Fiber substrate adhesion and coatings by contact metathesis polymerization |
| US6368346B1 (en) * | 1999-06-03 | 2002-04-09 | American Medical Systems, Inc. | Bioresorbable stent |
| US6388043B1 (en) * | 1998-02-23 | 2002-05-14 | Mnemoscience Gmbh | Shape memory polymers |
| US20020137864A1 (en) * | 2001-01-24 | 2002-09-26 | Tong Tat Hung | Shape memory styrene copolymer |
| US6530950B1 (en) * | 1999-01-12 | 2003-03-11 | Quanam Medical Corporation | Intraluminal stent having coaxial polymer member |
| US6538089B1 (en) * | 1999-02-05 | 2003-03-25 | Forskarpatent I Syd Ab | Gels with a shape memory |
| US20030060793A1 (en) * | 2001-07-24 | 2003-03-27 | Topolkaraev Vasily A. | Disposable products having humidity activated materials with shape-memory |
| US20030060530A1 (en) * | 2001-07-24 | 2003-03-27 | Topolkaraev Vasily A. | Methods of making humidity activated materials having shape-memory |
| US6596818B1 (en) * | 1996-10-08 | 2003-07-22 | Alan M. Zamore | Irradiation conversion of thermoplastic to thermoset polymers |
| US20030147046A1 (en) * | 2002-02-06 | 2003-08-07 | Shadduck John H. | Adaptive optic lens system and method of use |
| US20030191276A1 (en) * | 2002-02-26 | 2003-10-09 | Mnemoscience Gmbh | Polymeric networks |
| US6679605B2 (en) * | 2000-05-22 | 2004-01-20 | Medennium, Inc. | Crystalline polymeric compositions for ophthalmic devices |
| US20040015187A1 (en) * | 2002-04-18 | 2004-01-22 | Mnemoscience Corporation | Biodegradable shape memory polymeric sutures |
| US20040014929A1 (en) * | 2002-04-18 | 2004-01-22 | Mnemoscience Gmbh | Polyester urethanes |
| US20040015261A1 (en) * | 2000-08-28 | 2004-01-22 | Hofmann Gregory J. | Shape memory polymer or alloy ophthalmic lens mold and methods of forming ophthalmic products |
| US20040024098A1 (en) * | 2002-07-30 | 2004-02-05 | Mather Patrick T. | Nonionic telechelic polymers incorporating polyhedral oligosilsesquioxane (POSS) and uses thereof |
| US20040024143A1 (en) * | 2002-04-18 | 2004-02-05 | Mnemoscience Gmbh | Interpenetrating networks |
| US20040030062A1 (en) * | 2002-05-02 | 2004-02-12 | Mather Patrick T. | Castable shape memory polymers |
| US20040122174A1 (en) * | 2002-10-11 | 2004-06-24 | Mather Patrick T. | Blends of amorphous and semicrystalline polymers having shape memory properties |
| US20040122184A1 (en) * | 2002-10-11 | 2004-06-24 | Mather Patrick T. | Crosslinked polycyclooctene |
| US20050010275A1 (en) * | 2002-10-11 | 2005-01-13 | Sahatjian Ronald A. | Implantable medical devices |
| US6858680B2 (en) * | 1999-07-20 | 2005-02-22 | Aortech Biomaterials Pty Ltd | Shape memory polyurethane or polyurethane-urea polymers |
| US20050216074A1 (en) * | 2002-10-11 | 2005-09-29 | Sahatjian Ronald A | Implantable medical devices |
| US7198639B2 (en) * | 2002-09-13 | 2007-04-03 | Bausch & Lomb Incorporated | Polysilsesquioxane containing polymeric compositions |
| US20070290167A1 (en) * | 2004-08-27 | 2007-12-20 | Mather Patrick T | Crosslinked liquid crystalline polymer, method for the preparation thereof, and articles derived therefrom |
Family Cites Families (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0422693B1 (en) | 1985-01-04 | 1995-06-21 | Thoratec Laboratories Corporation | Method for making an article with shape-memory properties and some of the thus obtained articles |
| DE3817624A1 (en) | 1988-05-25 | 1989-11-30 | Bayer Ag | MIXTURES MADE FROM METASTABLE AMORPHES, PARTIAL KIRSTALLINE THERMOPLASTICS |
| CA2002408A1 (en) | 1988-11-11 | 1990-05-11 | Shoji Suzuki | Structural material and its application |
| KR930001716B1 (en) | 1989-02-28 | 1993-03-12 | 다이낀 고오교오 가부시끼가이샤 | Polymer material having shape memory characteristics |
| EP0674683B1 (en) | 1992-12-21 | 1997-10-15 | Raychem Corporation | Polymeric blends |
| NL9400519A (en) | 1994-03-31 | 1995-11-01 | Rijksuniversiteit | Intravascular polymeric stent. |
| GB9803770D0 (en) | 1998-02-23 | 1998-04-15 | Unilever Plc | Detergent compositions |
| US6312461B1 (en) | 1998-08-21 | 2001-11-06 | John D. Unsworth | Shape memory tubular stent |
| AU763085B2 (en) | 1998-11-12 | 2003-07-10 | Takiron Co. Ltd. | Shape-memory, biodegradable and absorbable material |
| CA2372746C (en) | 1999-05-24 | 2012-10-02 | California Institute Of Technology | Imidazolidine-based metal carbene metathesis catalysts |
| CN1214034C (en) | 1999-08-04 | 2005-08-10 | 杂混复合塑料公司 | Process for the formation of polyhedral oligomeric silsesquioxanes |
| AU2001263946A1 (en) | 2000-05-31 | 2001-12-11 | Mnemoscience Gmbh | Shape memory thermoplastics and polymer networks for tissue engineering |
| US6607553B1 (en) | 2000-11-17 | 2003-08-19 | B. Braun Medical, Inc. | Method for deploying a thermo-mechanically expandable stent |
| WO2002083786A1 (en) | 2001-04-11 | 2002-10-24 | Cheong Seok Hong | Shape memory rubber composition |
| DE10152146A1 (en) | 2001-10-25 | 2003-05-22 | Ticona Gmbh | Process for reducing the toughness of molded plastic parts to be machined and their use |
| DE10215858A1 (en) | 2002-04-10 | 2004-03-18 | Mnemoscience Gmbh | Process for hair treatment with shape memory polymers |
| US20050244353A1 (en) | 2002-04-10 | 2005-11-03 | Mnemoscience Gmbh | Method for achieving shape memory effects on hair by combining shape memory polymers with cationic active ingredients |
| DK1519713T3 (en) | 2002-07-10 | 2011-01-10 | Geesthacht Gkss Forschung | Active ingredient release systems based on biodegradable or biocompatible polymers with a shape memory effect |
| WO2004011525A1 (en) | 2002-07-30 | 2004-02-05 | University Of Connecticut | Nonionic telechelic polymers incorporating polyhedral oligosilsesquioxane (poss) and uses thereof |
| CA2532548A1 (en) | 2003-07-18 | 2005-02-03 | Boston Scientific Limited | Medical devices |
| JP2005102953A (en) | 2003-09-30 | 2005-04-21 | Tomii Kk | Orthodontic appliance |
-
2005
- 2005-04-21 US US11/111,388 patent/US7524914B2/en not_active Expired - Fee Related
-
2006
- 2006-04-11 JP JP2008507724A patent/JP2008537010A/en active Pending
- 2006-04-11 WO PCT/US2006/013797 patent/WO2006115799A1/en active Application Filing
- 2006-04-11 CA CA002605497A patent/CA2605497A1/en not_active Abandoned
- 2006-04-11 AU AU2006240293A patent/AU2006240293B2/en not_active Ceased
- 2006-04-11 EP EP06749989A patent/EP1907434A1/en not_active Withdrawn
-
2009
- 2009-03-13 US US12/403,410 patent/US20090253842A1/en not_active Abandoned
Patent Citations (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3459725A (en) * | 1964-01-17 | 1969-08-05 | Montedison Spa | High molecular weight unsaturated hydrocarbon homopolymers and process for preparing them |
| US3563973A (en) * | 1965-03-04 | 1971-02-16 | Radiation Applic Inc | Articles with polymeric memory and method of constructing same |
| US3383336A (en) * | 1966-06-27 | 1968-05-14 | Kuyama Hiroshi | Resinous insoluble reaction products |
| US5506300A (en) * | 1985-01-04 | 1996-04-09 | Thoratec Laboratories Corporation | Compositions that soften at predetermined temperatures and the method of making same |
| US4612241A (en) * | 1985-06-12 | 1986-09-16 | E. I. Du Pont De Nemours And Company | Impact resistant composites with elastomeric fibers |
| US5145935A (en) * | 1988-09-30 | 1992-09-08 | Mitsubishi Jukogyo Kabushiki Kaisha | Shape memory polyurethane elastomer molded article |
| US5189110A (en) * | 1988-12-23 | 1993-02-23 | Asahi Kasei Kogyo Kabushiki Kaisha | Shape memory polymer resin, composition and the shape memorizing molded product thereof |
| US5282854A (en) * | 1989-10-11 | 1994-02-01 | Daikin Industries Ltd. | Fluorine-containing block copolymer and artificial lens comprising the same |
| US5147385A (en) * | 1989-11-01 | 1992-09-15 | Schneider (Europe) A.G. | Stent and catheter for the introduction of the stent |
| US5258020A (en) * | 1990-09-14 | 1993-11-02 | Michael Froix | Method of using expandable polymeric stent with memory |
| US5163952A (en) * | 1990-09-14 | 1992-11-17 | Michael Froix | Expandable polymeric stent with memory and delivery apparatus and method |
| US5607467A (en) * | 1990-09-14 | 1997-03-04 | Froix; Michael | Expandable polymeric stent with memory and delivery apparatus and method |
| US6248129B1 (en) * | 1990-09-14 | 2001-06-19 | Quanam Medical Corporation | Expandable polymeric stent with memory and delivery apparatus and method |
| US5769883A (en) * | 1991-10-04 | 1998-06-23 | Scimed Life Systems, Inc. | Biodegradable drug delivery vascular stent |
| US5665822A (en) * | 1991-10-07 | 1997-09-09 | Landec Corporation | Thermoplastic Elastomers |
| US5395882A (en) * | 1992-09-24 | 1995-03-07 | Rohm Gmbh Chemische Fabrik | Light-scattering polystyrene molding compounds, and molded articles produced therefrom |
| US5964744A (en) * | 1993-01-04 | 1999-10-12 | Menlo Care, Inc. | Polymeric medical device systems having shape memory |
| US5674242A (en) * | 1995-06-06 | 1997-10-07 | Quanam Medical Corporation | Endoprosthetic device with therapeutic compound |
| US5954744A (en) * | 1995-06-06 | 1999-09-21 | Quanam Medical Corporation | Intravascular stent |
| US5880240A (en) * | 1996-04-03 | 1999-03-09 | Arakawa Chemical Industries, Ltd. | Alkyl-containing porous resin, process for its preparation and its use |
| US5889118A (en) * | 1996-06-03 | 1999-03-30 | Minnesota Mining And Manufacturing Company | Thermomorphic "smart" pressure sensitive adhesives |
| US5910357A (en) * | 1996-07-12 | 1999-06-08 | Nitto Denko Corporation | Separation membrane and method of producing the same, and shape memory polymer composition |
| US5908918A (en) * | 1996-07-17 | 1999-06-01 | Chronopol, Inc. | Impact modified polylactide |
| US5955559A (en) * | 1996-09-17 | 1999-09-21 | Shell Oil Company | Cast polyurethane elastomers containing low polarity amine curing agents |
| US5900444A (en) * | 1996-10-08 | 1999-05-04 | Zamore; Alan | Irradiation conversion of thermoplastic to thermoset polyurethane |
| US6596818B1 (en) * | 1996-10-08 | 2003-07-22 | Alan M. Zamore | Irradiation conversion of thermoplastic to thermoset polymers |
| US6024764A (en) * | 1997-08-19 | 2000-02-15 | Intermedics, Inc. | Apparatus for imparting physician-determined shapes to implantable tubular devices |
| US6395038B1 (en) * | 1997-08-19 | 2002-05-28 | Intermedics Inc. | Apparatus for imparting physician-determined shapes to implantable tubular devices |
| US6388043B1 (en) * | 1998-02-23 | 2002-05-14 | Mnemoscience Gmbh | Shape memory polymers |
| US6160084A (en) * | 1998-02-23 | 2000-12-12 | Massachusetts Institute Of Technology | Biodegradable shape memory polymers |
| US6720402B2 (en) * | 1998-02-23 | 2004-04-13 | Mnemoscience Gmbh | Shape memory polymers |
| US6156842A (en) * | 1998-03-11 | 2000-12-05 | The Dow Chemical Company | Structures and fabricated articles having shape memory made from α-olefin/vinyl or vinylidene aromatic and/or hindered aliphatic vinyl or vinylidene interpolymers |
| US6217609B1 (en) * | 1998-06-30 | 2001-04-17 | Schneider (Usa) Inc | Implantable endoprosthesis with patterned terminated ends and methods for making same |
| US6183248B1 (en) * | 1998-11-30 | 2001-02-06 | Muhammad Chishti | System and method for releasing tooth positioning appliances |
| US6705861B2 (en) * | 1998-11-30 | 2004-03-16 | Align Technology, Inc. | System and method for releasing tooth positioning appliances |
| US6390812B1 (en) * | 1998-11-30 | 2002-05-21 | Align Technology, Inc. | System and method for releasing tooth positioning appliances |
| US6485298B2 (en) * | 1998-11-30 | 2002-11-26 | Align Technology, Inc. | System and method for releasing tooth positioning appliances |
| US20020015519A1 (en) * | 1998-12-11 | 2002-02-07 | Lord Corporation | Fiber substrate adhesion and coatings by contact metathesis polymerization |
| US6530950B1 (en) * | 1999-01-12 | 2003-03-11 | Quanam Medical Corporation | Intraluminal stent having coaxial polymer member |
| US6538089B1 (en) * | 1999-02-05 | 2003-03-25 | Forskarpatent I Syd Ab | Gels with a shape memory |
| US6368346B1 (en) * | 1999-06-03 | 2002-04-09 | American Medical Systems, Inc. | Bioresorbable stent |
| US6858680B2 (en) * | 1999-07-20 | 2005-02-22 | Aortech Biomaterials Pty Ltd | Shape memory polyurethane or polyurethane-urea polymers |
| US6086204A (en) * | 1999-09-20 | 2000-07-11 | Magnante; Peter C. | Methods and devices to design and fabricate surfaces on contact lenses and on corneal tissue that correct the eye's optical aberrations |
| US20020007222A1 (en) * | 2000-04-11 | 2002-01-17 | Ashvin Desai | Method and apparatus for supporting a body organ |
| US6679605B2 (en) * | 2000-05-22 | 2004-01-20 | Medennium, Inc. | Crystalline polymeric compositions for ophthalmic devices |
| US20040015261A1 (en) * | 2000-08-28 | 2004-01-22 | Hofmann Gregory J. | Shape memory polymer or alloy ophthalmic lens mold and methods of forming ophthalmic products |
| US20020137864A1 (en) * | 2001-01-24 | 2002-09-26 | Tong Tat Hung | Shape memory styrene copolymer |
| US20030060530A1 (en) * | 2001-07-24 | 2003-03-27 | Topolkaraev Vasily A. | Methods of making humidity activated materials having shape-memory |
| US20030060793A1 (en) * | 2001-07-24 | 2003-03-27 | Topolkaraev Vasily A. | Disposable products having humidity activated materials with shape-memory |
| US20030147046A1 (en) * | 2002-02-06 | 2003-08-07 | Shadduck John H. | Adaptive optic lens system and method of use |
| US20030191276A1 (en) * | 2002-02-26 | 2003-10-09 | Mnemoscience Gmbh | Polymeric networks |
| US20040015187A1 (en) * | 2002-04-18 | 2004-01-22 | Mnemoscience Corporation | Biodegradable shape memory polymeric sutures |
| US20040024143A1 (en) * | 2002-04-18 | 2004-02-05 | Mnemoscience Gmbh | Interpenetrating networks |
| US20040014929A1 (en) * | 2002-04-18 | 2004-01-22 | Mnemoscience Gmbh | Polyester urethanes |
| US6852825B2 (en) * | 2002-04-18 | 2005-02-08 | Mnemoscience Gmbh | Polyester urethanes |
| US20060041089A1 (en) * | 2002-05-02 | 2006-02-23 | Mather Patrick T | Castable shape memory polymers |
| US20040030062A1 (en) * | 2002-05-02 | 2004-02-12 | Mather Patrick T. | Castable shape memory polymers |
| US20040024098A1 (en) * | 2002-07-30 | 2004-02-05 | Mather Patrick T. | Nonionic telechelic polymers incorporating polyhedral oligosilsesquioxane (POSS) and uses thereof |
| US7067606B2 (en) * | 2002-07-30 | 2006-06-27 | University Of Connecticut | Nonionic telechelic polymers incorporating polyhedral oligosilsesquioxane (POSS) and uses thereof |
| US7198639B2 (en) * | 2002-09-13 | 2007-04-03 | Bausch & Lomb Incorporated | Polysilsesquioxane containing polymeric compositions |
| US20040122184A1 (en) * | 2002-10-11 | 2004-06-24 | Mather Patrick T. | Crosslinked polycyclooctene |
| US20050216074A1 (en) * | 2002-10-11 | 2005-09-29 | Sahatjian Ronald A | Implantable medical devices |
| US20050010275A1 (en) * | 2002-10-11 | 2005-01-13 | Sahatjian Ronald A. | Implantable medical devices |
| US7091297B2 (en) * | 2002-10-11 | 2006-08-15 | The University Of Connecticut | Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments |
| US20040122174A1 (en) * | 2002-10-11 | 2004-06-24 | Mather Patrick T. | Blends of amorphous and semicrystalline polymers having shape memory properties |
| US20070290167A1 (en) * | 2004-08-27 | 2007-12-20 | Mather Patrick T | Crosslinked liquid crystalline polymer, method for the preparation thereof, and articles derived therefrom |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090042999A1 (en) * | 2007-08-09 | 2009-02-12 | Samsung Electronics Co., Ltd. | Composition for polyurethane foam, polyurethane foam made from the composition, and method for preparing polyurethane foam |
| US20110092652A1 (en) * | 2009-10-20 | 2011-04-21 | Voit Walter E | Shape memory polymers and process for preparing |
| WO2011049879A1 (en) * | 2009-10-20 | 2011-04-28 | Georgia Tech Research Corporation | Shape memory polymers and process for preparing |
| US8299191B2 (en) | 2009-10-20 | 2012-10-30 | Georgia Tech Research Corp. | Shape memory polymers and process for preparing |
| DE102010040762B4 (en) * | 2010-09-14 | 2017-08-03 | Technische Hochschule Wildau | Inorganic-organic polymeric hybrid networks, process for their preparation and their use |
| DE102010040762A1 (en) * | 2010-09-14 | 2012-03-15 | Technische Hochschule Wildau | Inorganic-organic polymer hybrid network, useful for preparing shape memory polymer, comprises hydroxyl group-containing thermoplastically processable polymer, nanoscale metal compounds, and reactive compounds |
| CN102617823A (en) * | 2012-03-29 | 2012-08-01 | 合肥工业大学 | Process for preparing hydroxyl polyhedral oligomeric silsesquioxane modified polyurethane |
| US8815054B2 (en) * | 2012-10-05 | 2014-08-26 | The Procter & Gamble Company | Methods for making fibrous paper structures utilizing waterborne shape memory polymers |
| US8980057B2 (en) | 2012-10-05 | 2015-03-17 | The Procter & Gamble Company | Fibrous paper structures utilizing waterborne shape memory polymers |
| US10563004B2 (en) | 2015-03-27 | 2020-02-18 | Basf Se | Memory foam based on thermoplastic polyurethane |
| US10407574B2 (en) * | 2016-10-20 | 2019-09-10 | Samsung Electronics Co., Ltd. | Composition with self-healing property, film with self-healing property and device including the film |
| US11041075B2 (en) | 2016-10-20 | 2021-06-22 | Samsung Electronics Co., Ltd. | Self-healing composition, self-healing film, and device including the self-healing film |
| CN109867947A (en) * | 2017-12-01 | 2019-06-11 | 财团法人纺织产业综合研究所 | Particles and yarn |
| CN111690111A (en) * | 2020-07-30 | 2020-09-22 | 中国科学院兰州化学物理研究所 | Comb type polymer and preparation method and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US7524914B2 (en) | 2009-04-28 |
| JP2008537010A (en) | 2008-09-11 |
| AU2006240293B2 (en) | 2011-02-24 |
| AU2006240293A1 (en) | 2006-11-02 |
| US20050245719A1 (en) | 2005-11-03 |
| CA2605497A1 (en) | 2006-11-02 |
| EP1907434A1 (en) | 2008-04-09 |
| WO2006115799A1 (en) | 2006-11-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7524914B2 (en) | Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments | |
| US7091297B2 (en) | Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments | |
| JP5600062B2 (en) | High modulus polyurethane and polyurethane / urea compositions | |
| Rueda-Larraz et al. | Synthesis and microstructure–mechanical property relationships of segmented polyurethanes based on a PCL–PTHF–PCL block copolymer as soft segment | |
| Heijkants et al. | Uncatalyzed synthesis, thermal and mechanical properties of polyurethanes based on poly (ε-caprolactone) and 1, 4-butane diisocyanate with uniform hard segment | |
| EP1687378B1 (en) | Preparation of supramolecular polymers containing quadruple hydrogen bonding units in the polymer backbone | |
| Ojha et al. | Syntheses and characterization of novel biostable polyisobutylene based thermoplastic polyurethanes | |
| US20070088135A1 (en) | Blends with shape memory characteristics | |
| US20060293487A1 (en) | Polyurethanes, polyurethaneureas and polyureas and use thereof | |
| CN108264623B (en) | Polyester type polyurethane shape memory material and preparation method thereof | |
| EP2066719B1 (en) | Polylactide-urethane copolymers | |
| WO2011133847A2 (en) | Polyhedral oligomeric silsesquioxane polyurethanes | |
| KR20190022787A (en) | Thermoplastic polyurethane with reduced tack | |
| EP3978569A1 (en) | Polymer composition, molded body and nerve regeneration inducing tube | |
| Pusztai et al. | The effect of some disiloxane chain extenders on the thermal and mechanical properties of cross-linked poly (siloxane-urethane) s. | |
| Kultys et al. | Polymers containing sulfur in the side chain | |
| Lipik et al. | Synthesis of biodegradable thermoplastic elastomers (BTPE) based on epsilon-caprolactone | |
| US20060122367A1 (en) | Process for preparing biocompatible polyurea |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |